1. Introdução
Sobre o que é este texto
Se uma pessoa ouve sobre uma "simulação da realidade", é mais provável que ela faça várias obras de ficção científica (como Matrix, The Dark City ou Zero Theorems) ou jogos de computador. No caso de pessoas cujas cabeças estão obstruídas com um diploma de engenharia, pacotes como o
KOMPAS-3D AutoCAD, o Solid Edge ou o NX podem aparecer. Uma pessoa que está ouvindo ciência provavelmente se lembrará de qualquer
modelagem de várias engenhocas espaciais .
Mas há mais um nível de Realidade, que será esquecido imerecidamente: aquele em que toda a química ocorre é o nível de átomos e moléculas. Também pode ser simulado com êxito em um computador. Como a mecânica quântica é responsável por tudo nesta seção da Realidade, esses cálculos são freqüentemente chamados de química quântica. E falaremos sobre sua conexão com a realidade estudada por métodos experimentais.
Este texto será sobre as coisas mais elementares. Porém, a prática de ler revistas científicas e ouvir vários relatórios mostra que isso deve ser constantemente lembrado.
O texto foi projetado para pessoas que entendem e / ou estão interessadas em como os átomos e moléculas vivem.Retirado de xkcd.comBreve históricoPor acaso, um colega, infelizmente, trabalhou na ciência russa, me convidou para dar uma palestra em seu curso especial para 2 pessoas em uma das conhecidas universidades físicas da Rússia. Mas, por uma estranha coincidência, ela foi transferida para uma conferência de estudantes realizada em paralelo ... Lá, ela não despertou muito interesse entre os alunos, e eu senti muito, muito pelo material, então decidi enviar um spam a Habr um pouco, tentando transformar a palestra educacional em um artigo popular de ciência.
Métodos físicos para estudar a vida das moléculas
Sabemos pelas aulas de química e física das escolas que todas as substâncias são compostas por átomos, moléculas, íons ou combinações dos mesmos. E parece que sabemos até que tipo de vida eles vivem. Mas essas informações devem ter suas próprias fontes confiáveis (métodos de pesquisa), e realmente são.
Existem muitas, muitas maneiras de espionar a vida dos átomos. Aqueles que desejam, por exemplo, podem se familiarizar com alguns deles em mais detalhes nos livros clássicos
- Pentin Yu.A., Vilkov L.V. Métodos de pesquisa física em química. - M. Mir, 2006,
- Drago R. Métodos físicos em química. - M .: Mir, 1981.
Porém, de maneira grosseira e fácil, três grupos principais de métodos se destacam:
- métodos espectroscópicos
- métodos de difração
- vários métodos de microscopia (não importa, translúcido ou digitalizado, para nós isso não é essencial agora).
Não se fala sobre o último, mas suas ferramentas não são menos importantes que as duas primeiras.
Por que não haverá conversa sobre microscopia(Simplesmente não evito a palavra em microscopia)
Métodos espectroscópicos para o estudo da matéria
Esse poderoso grupo de métodos nos fornece muitas, muitas coisas: desde a busca e determinação de moléculas no meio interestelar e em outros planetas até a verificação banal de explosivos no aeroporto.
O princípio geral dos métodos espectrais
Ao falar sobre espectroscopia, geralmente se refere ao seguinte princípio geral de operação.
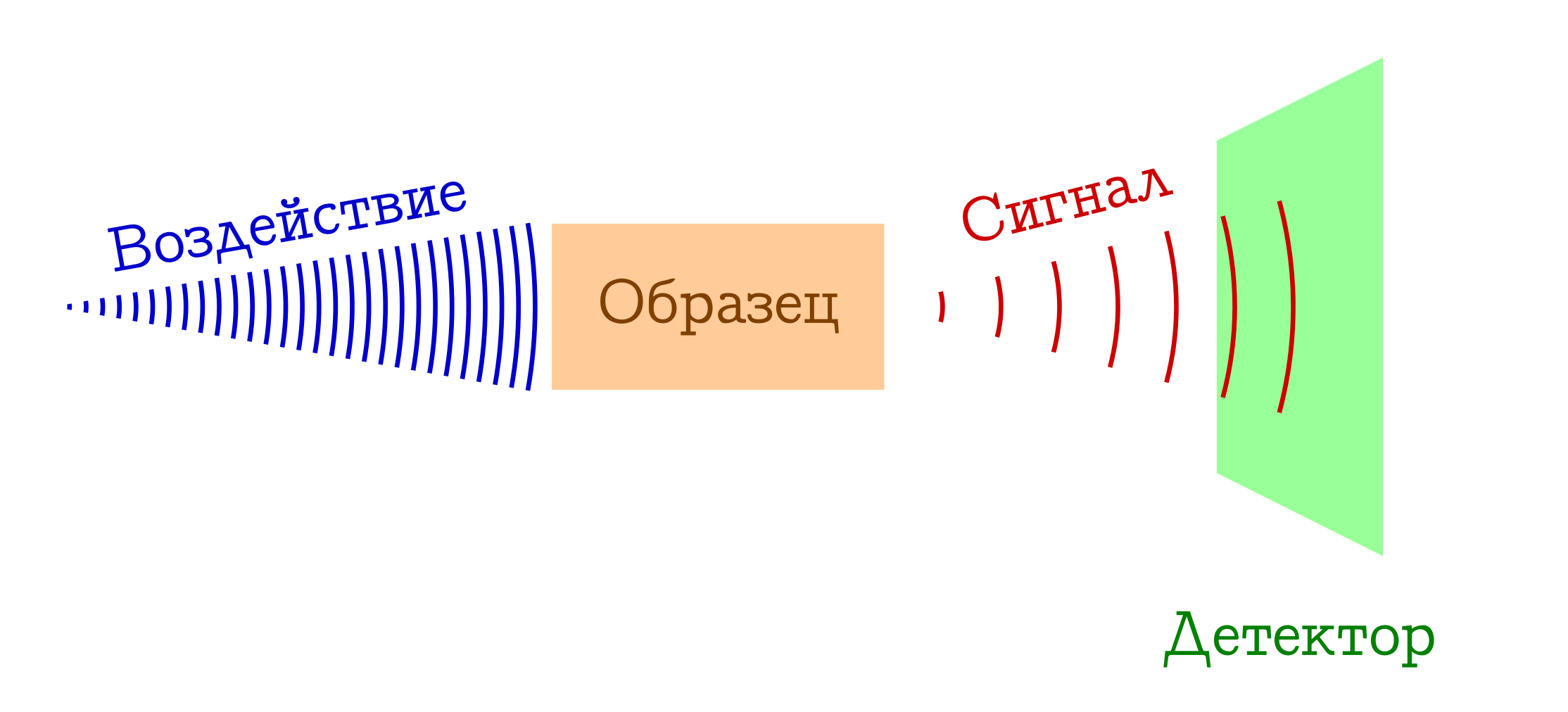
O esquema geral dos métodos espectrais para estudar substâncias
- Temos algo com o qual nós (por exemplo, uma lâmpada / laser / luz solar) agimos sobre a amostra de seu interesse. Na maioria das vezes, esse é um estudo eletromagnético, mas pode muito bem ser elétrons (por exemplo, em espectroscopia de massa com ionização por impacto de elétrons) ou um coquetel de tudo o que é possível e impossível com o plasma (por exemplo, em espectroscopia de chama , tão amada por crianças em idade escolar e estudantes de faculdade de química). De uma forma ou de outra, algo deve funcionar em nossa amostra.
- Quando exposto a uma amostra, acontece algo que muda de estado. Isso pode ser uma transição para algum tipo de nível excitado (em qualquer espectrofotometria ou espectroscopia Raman), ou mesmo o colapso do sistema molecular (como em espectros de massa ou espectroscopia de fotoelétrons ). Mas, de alguma forma, o padrão em algum momento deve ser diferente.
- ???
- LUCRO !!! Registramos um certo sinal (emitido ou absorvido) com essa alteração na amostra no nível molecular. Isso pode ser perda de fótons gastos na alteração da amostra (então temos espectroscopia de absorção), ou vice-versa, excesso de fótons emitidos após excitação preliminar da substância (espectroscopia de emissão), uma alteração no comprimento de onda dos fótons iniciais como resultado da interação com a substância (espectroscopia Raman, mais conhecido no exterior como
Ramenovskaya Ramanova ), ou fragmentos estupidamente das moléculas originais (como em espectros de massa ou espectroscopia de fotoelétrons ). Existem muitas opções - a essência é a mesma: há um sinal!
Como exemplo de tais métodos, você pode citar várias letras diferentes: RMN, ESR, MW, THz, IR, UV / Vis, XRF, MS, PES, EXAFS, XANES, etc. etc.
Todos (ou muitos deles) são familiares (ou deveriam ser familiares) para todos os químicos. Todos esses métodos são o arsenal padrão (longe de incompleto) de um pesquisador que se preze que lida com substâncias.
Intervalos espectrais e sua relação com a vida das moléculas
 Retirado de xkcd.com
Retirado de xkcd.comComo na esmagadora maioria dos casos a espectroscopia ainda está ligada à radiação eletromagnética, é lógico vincular as faixas do espectro eletromagnético a vários aspectos da vida molecular-atômica. Afinal, a frequência das ondas eletromagnéticas usadas na espectroscopia é uma espécie de "relógio" que permite detectar quanto tempo esse ou aquele processo em sistemas moleculares dura. Então, mudando essa frequência, você pode estudar (e até agir) em diferentes processos moleculares.
Então
- Do ponto de vista químico, nada de interessante acontece na faixa de comprimento de onda super longo, então você não consegue se lembrar.
- Com a frequência de rádio e microondas (e até radiação infravermelha de ondas longas, IR = IR), diferentes moléculas giram na fase gasosa: grandes e pesadas - na região de ondas de rádio (frequências mais baixas) e pequenas e leves - em IR (frequências mais altas).
- Na IR, no entanto, (principalmente) ocorrem várias vibrações moleculares: todos os movimentos conformacionais e outros movimentos não óbvios dentro das moléculas estão no IR de longo comprimento de onda, e vibrações de alongamento (alongamento - encurtamento dos comprimentos das ligações químicas) ocorrem no comprimento de onda curto (até 4000 cm –1 ).
- Bem, então vem o lugar do espectro, onde vivem várias transições eletrônicas (até a região de γ-quanta). Em frequências mais baixas (visível, UV = UV e raio X macio), principalmente transições associadas a elétrons de valência vivem.
Por que nós vemos?A propósito, é precisamente por causa das transições eletrônicas que podemos ver: aos nossos olhos (nos cones) existem estruturas que possuem uma
retina em sua composição. Quando um fóton visível é absorvido por essa molécula, uma ligação dupla quebra nele, o que leva à isomerização cis-trans. E é essa mudança que percebemos como o sinal primário, que é então transmitido ao nosso cérebro.
Mas com o aumento da energia de fótons (ou seja, com frequência crescente, como lembramos da fórmula de Planck E = h n u ) chegamos a camadas cada vez mais profundas da estrutura eletrônica, até descansarmos na faixa de raios-x até as conchas 1s finais ( ou, como são chamadas de raios-X, K ).
Assim, escolhendo o comprimento de onda certo da radiação eletromagnética, podemos observar mais detalhadamente um processo específico nas moléculas.
Métodos de difração de substâncias
Agora vamos falar um pouco sobre difração. O diagrama esquemático de tais experiências também é simples.
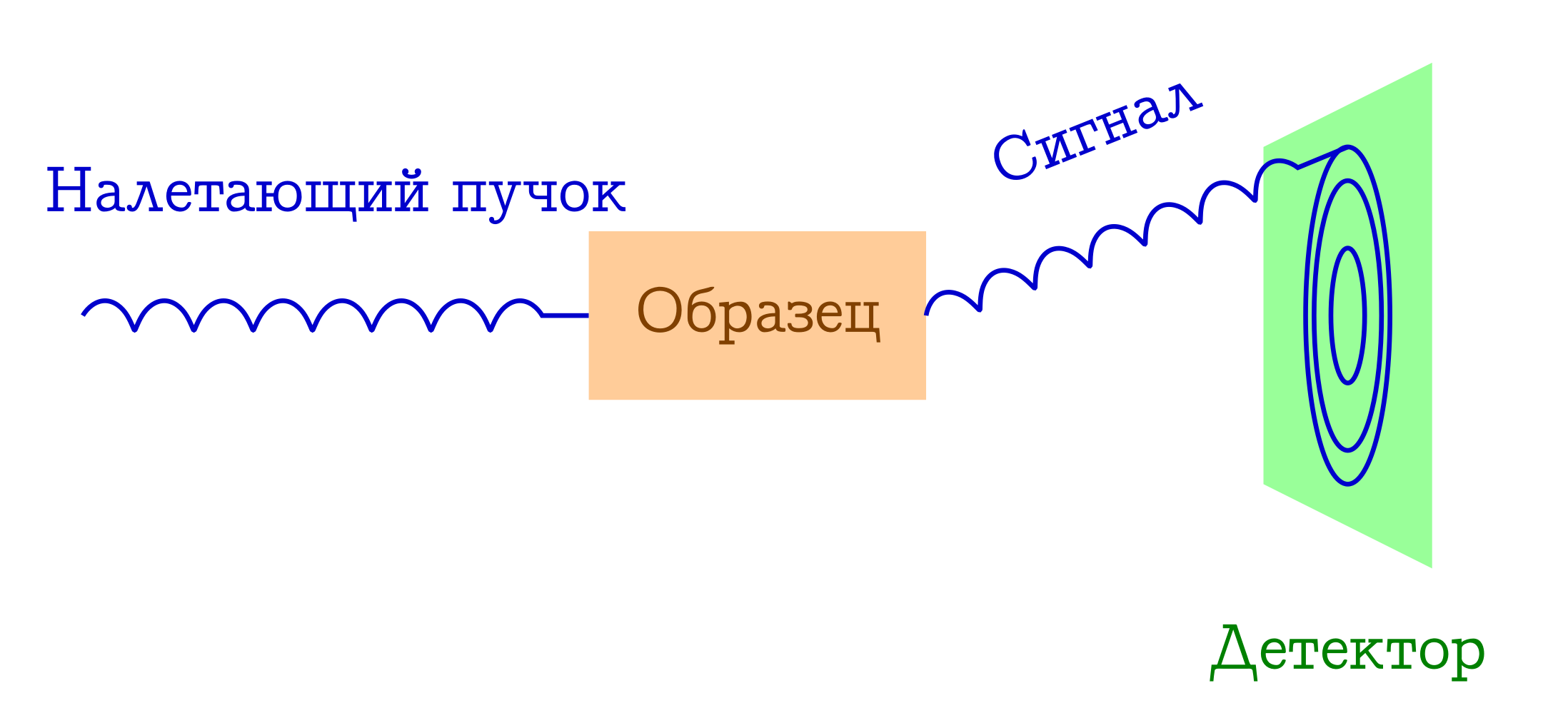
Esquema geral dos métodos de difração para o estudo de substâncias
- Um feixe de algumas partículas voa sobre a amostra. Na maioria das vezes, são fótons de raios X, elétrons ou nêutrons.
- Essas partículas, por diferentes mecanismos, espalham elasticamente os átomos na amostra de seu interesse (isto é, sem alterar o comprimento de onda e a fase da onda, elas simplesmente mudam a direção de seu voo). Nada acontece com a amostra dessas partículas incidentes: ele simplesmente não tem tempo para reagir a elas.
- As distâncias interatômicas servem como uma grade de difração para o feixe incidente; portanto, como resultado, veremos uma bela imagem de difração no detector.
A partir do último parágrafo, surge a condição para o comprimento de onda das partículas incidentes (λ): ela deve ser da mesma ordem ou menor que a ordem característica das distâncias interatômicas, de modo que λ típico para esses métodos é 1 - 0,01 Å.
Os principais tipos de erros ao comparar experimentos e cálculos teóricos
Como resultado, temos uma imagem muito interessante: na espectroscopia e na difração, observamos algum tipo de sinal esquerdo, que de alguma
forma indica
indiretamente o que realmente acontece no sistema molecular.
A analogia com a caverna platônicaEsta pintura lembra estranhamente o
mito da Caverna de Platão . Temos um certo mundo real de moléculas. Mas só vemos sombras dele na parede da caverna (detector), que são uma exibição incompleta de todas as coisas interessantes que acontecem neste nível de Realidade.
Mas, felizmente, às vezes podemos teoricamente calcular o sinal de interesse para nós (como, por exemplo, em microondas, espectroscopia de infravermelho ou UV / Vis), e às vezes podemos extrair do sinal observado as quantidades de interesse disponíveis para o cálculo químico quântico (por exemplo, distância entre átomos de uma molécula, momento dipolar, etc.). E aqui temos a chance de que o experimento numérico e o real possam se unir no estágio apaixonado da comparação entre si ... e aqui 4 tipos de erros podem ocorrer como padrão.
Atenção! O termo "erro" aqui não significa que o resultado da comparação esteja obviamente errado. Só que o motivo da comparação se torna muito instável e pantanoso, e um passo desleixado pode facilmente atrapalhar todo o trabalho.
- Diferentes condições do experimento e / ou cálculo (estado de agregação, temperatura, pressão, etc.). De repente, podemos começar a comparar sistemas diferentes entre si, por algum motivo, considerando-os iguais. Por exemplo, é óbvio que adicionar uma ou cinco colheres de chá de açúcar a uma xícara de chá levará ao mesmo sistema físico chamado “chá com açúcar”, mas as propriedades desse sistema serão muito diferentes. E pode ser facilmente medido. Por exemplo, com um termômetro (medindo a temperatura do chá imediatamente após a dissolução do açúcar) ou com a língua (um dos chamados métodos de análise organoléptica). Portanto, ao comparar os sistemas resultantes entre si (seja um copo de açúcar com chá ou seu modelo de computador), não devemos esquecer que as semelhanças têm seus limites e que, se reduzirmos a margem de erro para "semelhança", encontraremos as diferenças.
- Significado físico e / ou matemático diferente dos parâmetros (o parâmetro do significado físico no sentido usual pode até não existir). Aqui também tudo é simples: se compararmos duas quantidades com um nome semelhante, isso não significa que as quantidades tenham o mesmo significado físico. Por exemplo, a classificação de deputado entre toda a população da cidade vs. classificação apenas entre avós. Tanto essa como a classificação (o que quer que seja), esses números (ou o que é) podem até se correlacionar fortemente, mas o significado desses parâmetros ainda é diferente e essa diferença pode ser detectada.
- Erros "aleatórios" . Isso se refere a alguns erros sistemáticos dos quais o teórico do experimentador / simulador não está ciente ou a erros realmente aleatórios no experimento / cálculo que não podem ser controlados e / ou previstos. Em princípio, essas coisas podem se tornar objeto de investigação de vários efeitos sistemáticos interessantes.
ou apenas uma estimativa da relação S / N mais útil ("sinal para ruído"). - E o último erro padrão é o crescimento das mãos do experimentador / calculadora a partir do osso pélvico , ou seja, erros humanos comuns. Não há necessidade de investigar nada, basta verificar o trabalho ou repetir o experimento para encontrar e eliminar o batente correspondente.
Nada mais concreto pode ser dito sobre os dois últimos tipos de erros, mas sobre os dois primeiros e, se você usar um método de pesquisa específico, poderá dizer muitas coisas. Portanto, vamos nos concentrar neles. A ênfase principal neste caso será sobre as diferenças estruturais das moléculas.
Erro # 1. Diferenças nas propriedades moleculares sob diferentes condições
NaCl: quando não há erros
Por alguma razão, não ocorre a ninguém dizer que o único cristal de cloreto de sódio (NaCl), que é uma enorme molécula de íons Na
+ e Cl
- , e a molécula diatômica de NaCl, obtida por evaporação desse cristal em temperaturas loucas, têm um e o mesmo digamos a estrutura.
E mesmo se assumirmos que pelo menos as distâncias entre cloro e sódio (
r NaCl ) são as mesmas aqui e ali, o experimento nos colocará em prática:
Onde cometemos um erroDe fato, com essa comparação, permitimos a possibilidade do Erro # 2, mas está tudo bem aqui, se avaliarmos os erros dessa comparação, eles serão da ordem de 0,01 Å, que é significativamente menor que a diferença dos parâmetros comparados. I.e. isso não é um erro, mas um efeito real.
Como obter a distância entre o cátion de sódio e o ânion de cloro em um cristal de salObter a distância entre os átomos de uma molécula de NaCl diatômica a partir de dados experimentais também não é um procedimento tão complicado. Mas o problema é apenas que esse experimento é uma coisa complicada. Portanto, é mais fácil usar um banco de dados onde as distâncias requeridas já estão especificadas.
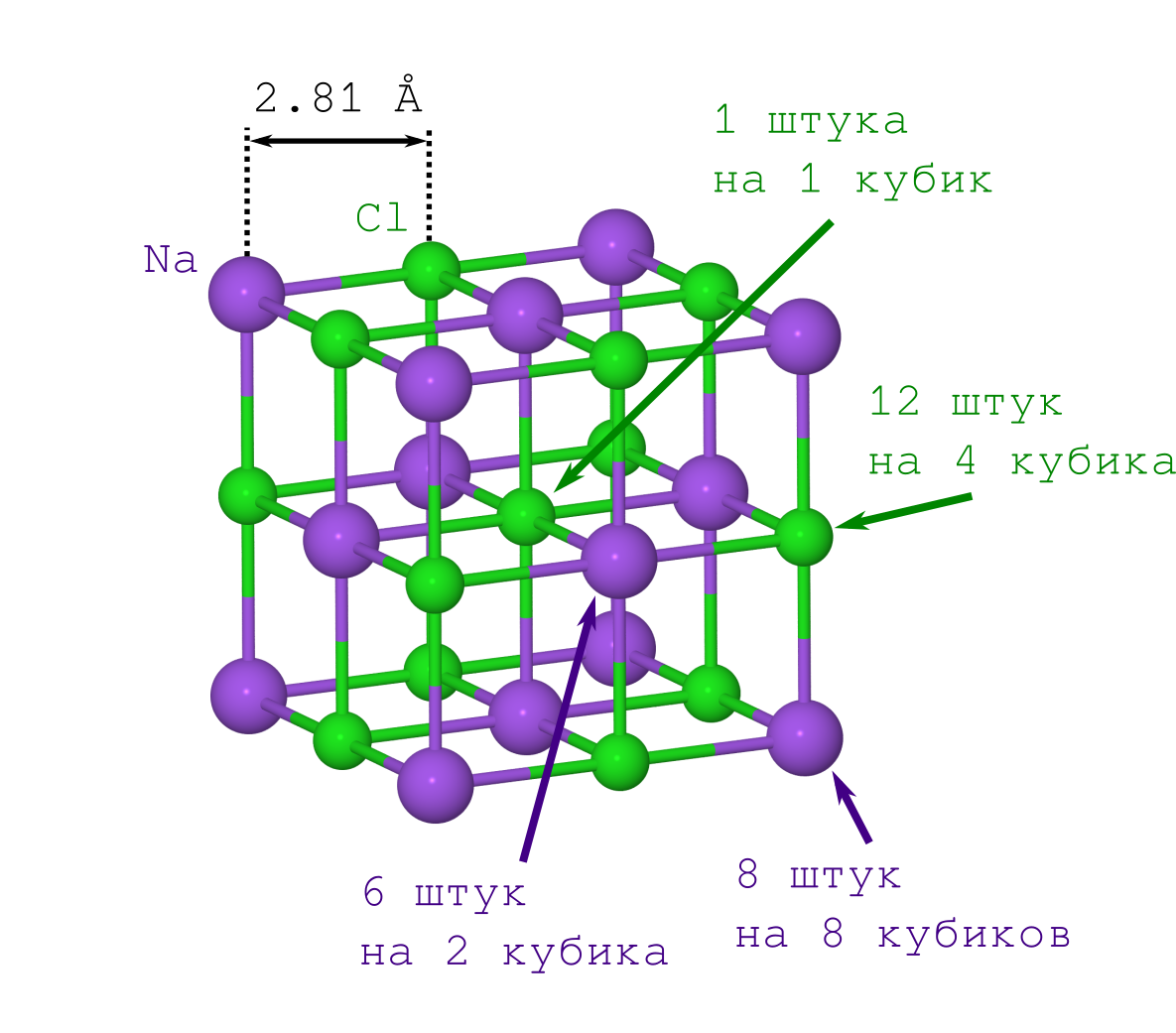
Mas, para obter a distância entre os átomos no cristal, apenas a densidade do sal cristalino da tabela ρ = 2,165 g / cm
3 é suficiente, o que pode
ser facilmente obtido na Wikipedia e medido em casa.
Para calcular a distância, precisamos:
- a densidade do cristal de NaCl (é),
- conhecimento da localização dos íons deste cristal.
Se você tivesse feito isso pela primeira vez (digamos, no começo do século 20), teria que se atormentar com o segundo ponto. Mas isso já é conhecido pelas pessoas modernas: a rede de NaCl tem a forma de um cubo no qual os íons Na
+ e Cl
- alternam entre si (veja a figura acima). Ao multiplicar o fragmento indicado do cristal ("copiar e colar" a peça especificada e ajustá-la à iteração anterior frente a frente), obtemos um cristal NaCl de qualquer tamanho e formato (minecraft) desejados.
Portanto, a densidade deste cubo deve ser a mesma que a de todo o cristal. Dado que a densidade é
r h o = f r a c m V (isto é, massa por volume), verifica-se que, conhecendo a massa e a expressão geométrica do volume, podemos calcular a distância entre os átomos.
O volume do cubo é óbvio: o comprimento da nervura é o dobro da distância Na - Cl (
L = 2 r m a t h r m N a C l ), o que significa que o volume desejado é
V = L 3 = 8 r m a t h r m N a C l 3 .
A massa não é tão simples. A maioria dos nossos átomos está nos vértices, bordas e faces do cubo, o que significa que eles pertencem simultaneamente a vários desses cubos. Isso deve ser levado em consideração nos cálculos.
Vamos começar com íons Na
+ . Temos apenas 2 tipos deles (veja o padrão de treliça de cristal):
- aqueles que estão nos vértices do cubo (existem tantos quanto os vértices do cubo, ou seja, 8, e eles estão simultaneamente em 8 cubos, portanto, você precisará dividir esse número por 8),
- aqueles que estão sobre os rostos (existem 6 deles e simultaneamente pertencem a 2 cubos).
Como resultado, obtemos que nosso cubo contém
8 cdot frac18+6 cdot frac12=4 íon sódio.
Agora sobre Cl
- . Existem também apenas 2 tipos deles (veja o padrão de treliça de cristal):
- aqueles que estão nas bordas do cubo (existem 12 deles e são de propriedade conjunta de 4 cubos),
- esse Cl - que está no centro do cubo, é um e pertence apenas ao nosso cubo.
Portanto, nosso cubo contém
12 cdot frac14+1 cdot frac11=4 íon cloro.
A composição do cristal, obviamente, corresponde à fórmula química do NaCl, mas a massa do nosso cubo é igual (não esqueça que as massas de átomos na tabela periódica são dadas em
unidades de massa atômica ):
m=4 cdot( underbraceM mathrmNa23 textam+ underbraceM mathrmCl35,5 textamu)=234 textamu=234 cdot1,66 cdot10−24 textr=3,88 cdot10−22 textg\.
Agora a partir da relação
rho= fracmV podemos fazer uma equação para o comprimento
r mathrmNaCl :
r mathrmNaCl3= left( fracm8 rho right) ,
que é facilmente resolvido:
r mathrmNaCl= left( fracm8 rho right)1/3= left( frac3,88 cdot10−22 [ textg]8 cdot2,17 [ textg/ textcm3] right)1/3=2,82 cdot10−8 [ textcm]=2,82 [ textÅ] .
A partir dos dados da cristalografia de raios-X 2.81 Å (por exemplo, de
Abrahams, SC; Bernstein, JL Precisão de um difratômetro automático. Medição dos fatores de estrutura do cloreto de sódio // Acta Crystallographica (1965) 18, 926-932 ) perdemos apenas 0,01 Å, o que é legal o suficiente.
Alguém pode pensar que a diferença de 0,45 Å é insignificante, mas esse é quase o raio de Bohr (0,52 Å), que é igual à distância mais provável do elétron e, pelos padrões atômicos, a diferença é enorme.
Por que NaCl na forma de uma molécula atômica 2 difere de um cristalTudo é muito simples aqui. A treliça de cristal infinito cria a possibilidade de "salto" irreversível para 3s
1 elétron de sódio por átomo de cloro, pois a diferença de carga resultante é compensada pela interação com os vizinhos.
3s
1 ( ), ,
«» :
Na:Cl↔Na+Cl−
() (), .
,
±1 , , .
NaCl (2.36 Å),
d=q⋅rNaCl=9.0 [] onde
q≥0 (
+q ,
−q )
, « » 0.21, ..
d=0.21⋅9.0=1.9 [qe⋅Å] , :
q=drNaCl=1.92.36=0.8 . «» 0.2 NaCl NaCl .
Ferroceno
Vale a pena passar dos cristais iônicos para os moleculares nos quais as moléculas são densamente compactadas, de modo que, de repente, torna-se possível comparar, e sem reservas.Mas a diferença não deve ser esquecida. E existe até um exemplo clássico sobre esse assunto: uma molécula de ferroceno .Essa é a conexão sanduíche mais simples. Nele, um átomo de ferro neutro (como uma costeleta) é ensanduichado entre dois anéis aromáticos de cinco membros (pães).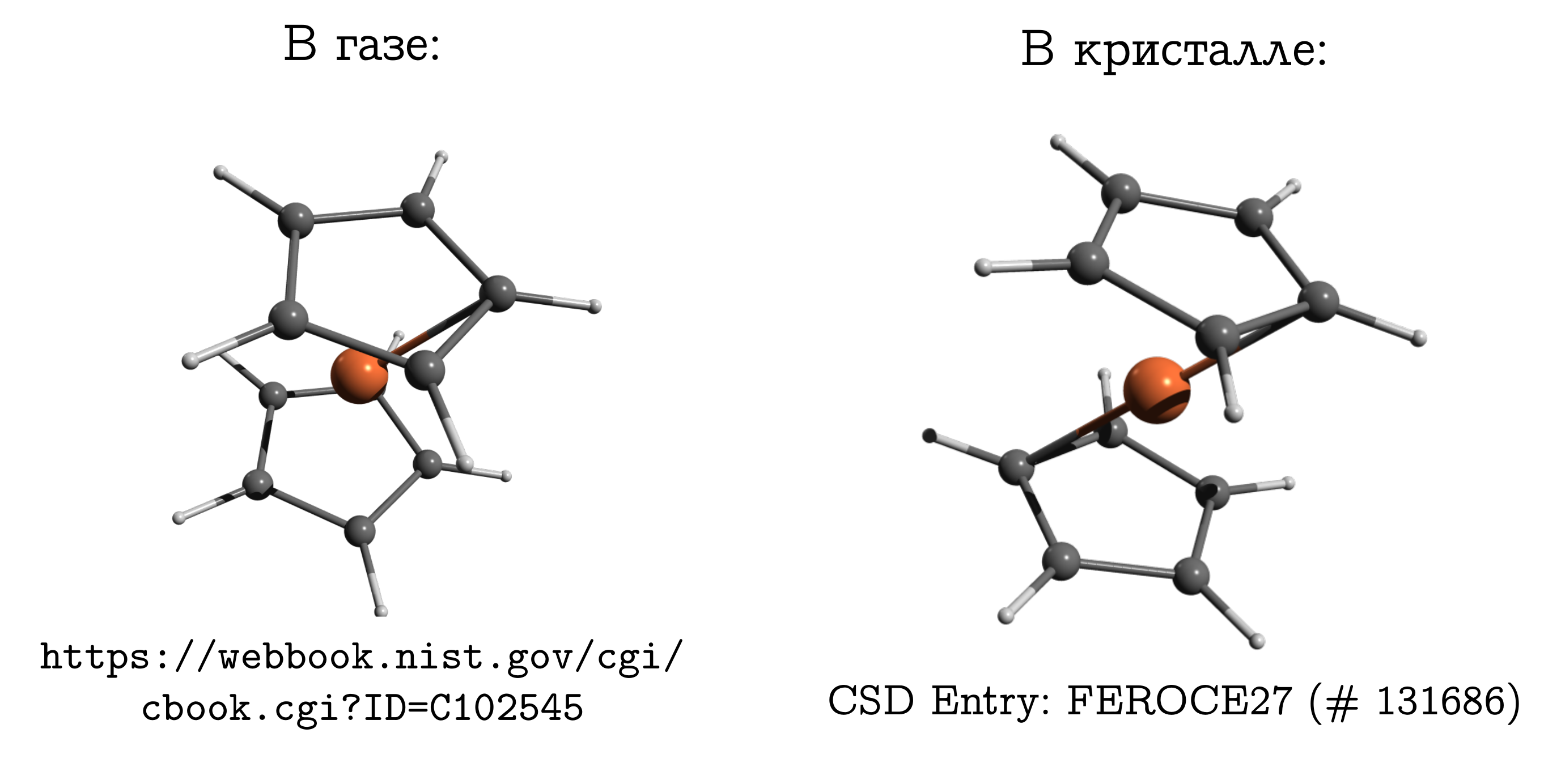 Essa molécula pode ser evaporada com bastante facilidade e descobrir que a chamada estrutura mais estável na fase gasosa é conformação obstruída. Nele, os carbonos e hidrogênios dos anéis superior e inferior são opostos (veja a figura acima), pois nesse caso as interações de dispersão são mais fortes entre essas partes da molécula, e a dispersão é sempre benéfica.Se pegarmos um cristal de ferroceno, verifica-se que as moléculas têm uma conformação estável diferente (que é chamada inibida por hidrocarbonetos), na qual o hidrogênio e o carbono de um anel estão acima / abaixo da ligação C - C do outro. Existem interações de dispersão entre moléculas, e uma similar, aparentemente inconveniente para uma estrutura de molécula, surge do fato de que é mais fácil para as moléculas se unirem apenas de uma forma desconfortável, e esse inconveniente pessoal é compensado pela interação entre si.
Essa molécula pode ser evaporada com bastante facilidade e descobrir que a chamada estrutura mais estável na fase gasosa é conformação obstruída. Nele, os carbonos e hidrogênios dos anéis superior e inferior são opostos (veja a figura acima), pois nesse caso as interações de dispersão são mais fortes entre essas partes da molécula, e a dispersão é sempre benéfica.Se pegarmos um cristal de ferroceno, verifica-se que as moléculas têm uma conformação estável diferente (que é chamada inibida por hidrocarbonetos), na qual o hidrogênio e o carbono de um anel estão acima / abaixo da ligação C - C do outro. Existem interações de dispersão entre moléculas, e uma similar, aparentemente inconveniente para uma estrutura de molécula, surge do fato de que é mais fácil para as moléculas se unirem apenas de uma forma desconfortável, e esse inconveniente pessoal é compensado pela interação entre si.Por que o ferroceno é tão diferente do etanoUma pessoa familiarizada com a química geralmente precisa se dominar para se lembrar da estrutura do ferroceno em um gás. Afinal, ele tem memórias de etano (C
2 H
6 ), nas quais a conformação mais estável é inibida (quando os hidrogênios de um pedaço de CH
3 ficam "entre" os hidrogênios de outro CH
3 ), porque nesta posição, a repulsão interatômica entre as camadas de elétrons dos hidrogênios é minimizada.

Adaptado de
www.chem.msu.su/rus/teaching/stereoE aqui toda a diferença está à distância. A forma padrão do potencial de interações de dispersão é o potencial de Lennard-Jones (esse, aliás, é um, não dois homens):
V mathrmLJ(r)= fracAr12− fracBr6
Nele, o primeiro termo é retirado da repulsão interatômica e o segundo da atração interatômica decorrente das flutuações da densidade eletrônica. Em geral, esse potencial se parece com isso:
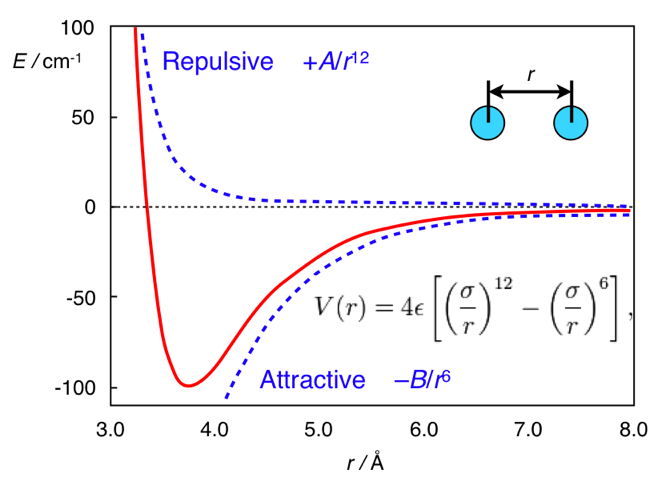
Potencial de Lennard-Jones. Adaptado de
chemistry.stackexchange.com/questions/34214/physical-significance-of-double-well-potential-in-quantum-bondingE no caso do etano, os átomos de hidrogênio estão muito próximos um do outro, portanto, estão (relativamente ao seu mínimo) no lado esquerdo da curva e são caracterizados por repulsão. No caso do ferroceno, entre os anéis existe uma camada de tamanho não doentio (um átomo de ferro), devido ao qual os anéis estão longe o suficiente para não sentir repulsa interatômica. E assim eles estão na parte certa (atraente) do potencial.
Histamina
No caso do ferroceno, vimos o chamado diferenças conformacionais: a molécula permaneceu a mesma (isto é, nenhuma ligação química foi quebrada ou formada) e sua forma mudou ligeiramente.
Mas as diferenças podem ser ainda mais fortes, por exemplo, se o chamado
transformações tautoméricas . A tautomerização é uma classe de reações químicas que ocorrem com tanta facilidade e rapidez que, como resultado, podemos ter simultaneamente vários isômeros de uma molécula, passando facilmente entre si. Esses isômeros são chamados tautômeros.
Um exemplo padrão disso: tautomerismo de ceto-enol em cetonas:

Na maioria das vezes, como neste exemplo, o tautomerismo está associado ao salto de um próton de um local quente para outro. E essas reações estão ligadas ao
efeito do
túnel , ao qual o hidrogênio, como o mais leve dos átomos, é mais suscetível.
Tais transformações químicas são características de muitas moléculas biológicas, por exemplo, as
bases nitrogenadas que compõem o DNA ou
açúcares .
Porém, durante a transição de sistema para sistema, as constantes de equilíbrio de tais reações geralmente mudam; portanto, em diferentes fases, podemos observar diferentes composições tautoméricas.
Um exemplo disso é a molécula de histamina (veja a figura abaixo).
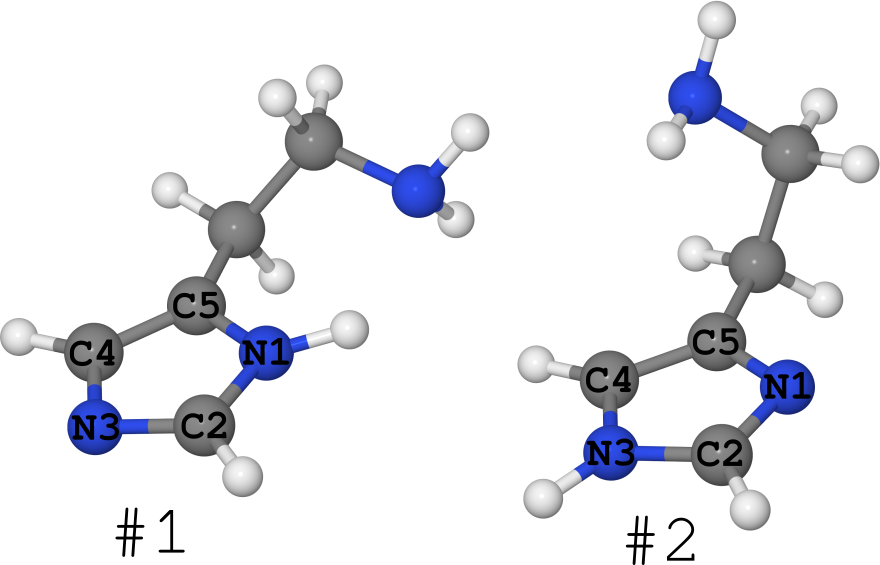
Existe na forma de 2 tautômeros (geralmente sou silencioso sobre o número de conformes, existem muitos):
- # 1, onde o hidrogênio fica no nitrogênio N1,
- # 2, onde o hidrogênio está no nitrogênio N3.
Aconteceu que para esta molécula são conhecidas suas estruturas em diferentes fases.
- No cristal, é completamente "congelado" na forma de # 1. (consulte o artigo DOI: 10.1021 / ja00796a011 e a estrutura do Cambridge Structures Bank sob o nome "HISTAN" e / ou número 1176642)
- Em soluções aquosas, essa molécula existe em ambas as formas e o tautômero nº 2 é visivelmente maior ( DOI: 10.1021 / ja027103x ).
- No gás, a histamina existe igualmente na forma 1 e 2 ( DOI: 10.1021 / ja980560m ).
I.e. fases diferentes contêm diferentes números de moléculas diferentes, o que significa que são sistemas diferentes.
Conclusão do erro # 1

A principal conclusão a ser tirada dos exemplos acima é a seguinte:
Ao comparar cálculos em uma fase com um experimento em outra, é preciso estar preparado para diferenças sistemáticas.
Isso não significa que não é necessário comparar: é necessário comparar, mas é apenas necessário ser mais crítico com as diferenças e / ou coincidências encontradas e avaliar esses efeitos, se possível.
Erro # 2. Parâmetros moleculares do "zoológico".
O segundo erro é descrito brevemente da seguinte maneira: se os parâmetros forem chamados de maneira semelhante, mas não idêntica, esses são parâmetros diferentes.
Para entender qual é a fonte dessa discordância entre teoria e experimento, será preciso analisar com mais detalhes os métodos experimentais padrão usados para obter parâmetros moleculares e os modelos que calculam quantidades semelhantes puramente a partir da teoria.
E aqui falaremos novamente apenas sobre estruturas.
Como obter estruturas moleculares experimentais
Para de alguma forma nos limitar, falaremos apenas sobre métodos para estudar a estrutura de moléculas únicas, ou seja, sobre a fase gasosa.
Temos duas fontes principais de informações:
- difração de elétrons de gás,
- espectroscopia de microondas.
Vamos abordar cada um desses métodos em mais detalhes.
Difração de elétrons a gás
O método é bastante antigo, tem origem nos anos 30 do século XX, quando os cientistas alemães Mark e Wirl conduziram os primeiros experimentos sobre a difração de elétrons por gás.
Poucas pessoas sabem, mas esse método de pesquisa está envolvido no recebimento de três prêmios Nobel em química.
3 nobres com entrada eletrográfica- Peter Debye em 1936 recebeu seu prêmio com a seguinte redação:
"[por seu trabalho em] estrutura molecular através de suas investigações sobre momentos dipolares e a difração de raios-X e elétrons em gases "
Esta é a única menção explícita da difração de elétrons gasosos nos méritos do laureado, e não sem razão. A equação básica de difração de elétrons para a intensidade de espalhamento molecular é denominada Debye.
de fato, a equação de DebyeIij(s)=gij frac sin(srij)rij
Aqui
Iij denota a intensidade de espalhamento de elétrons (ou raios-x ou outras partículas) por um par de
i- ésimo
j -
étimo átomo à distância
rij separado
s= frac2 pi lambda sin left( frac theta2 right) A coordenada de dispersão está associada ao ângulo de dispersão
theta e comprimento de onda das partículas
lambda e
g - a capacidade desse par de átomos de espalhar partículas difratantes.
E, apesar de qualquer coisa ser lembrada sobre essa física maravilhosa (o modelo de soluções iônicas , seu modelo para calcular a capacidade de calor dos cristais ), mas não a difração de elétrons, ele recebeu o principal prêmio científico (em particular) por isso.
- Linus Pauling em 1954. Sim, aquele que recebeu 2 prêmios Nobel pessoais
e até colocou o mundo inteiro em vitamina C , o Great Pauling. Quando ele estava trabalhando em Kaltekh, em particular, ele estava envolvido em difração de elétrons a gás (veja, por exemplo, DOI: 10.1021 / ja01873a047 ). E, é claro, o conhecimento da química estrutural das moléculas livres o ajudou a criar a famosa teoria das ligações químicas (mas não vamos subestimar sua grande experiência cristalográfica aqui). - Odd Hassel, Laureado de 1969. Ele recebeu seu 1/2 Prêmio Nobel pela descoberta do equilíbrio conformacional. Ele fez isso com base em um estudo de difração de elétrons de ciclo-hexano. Esta molécula existe na forma de duas conformações: uma cadeira (cadeira) e um banheiro (na tradição inglesa - um barco, barco).

A partir daqui: www.shapeways.com/product/N5FE298DS/cyclohexane-2-molecules-boat-and-chair-form
Essas opções para o arranjo dos átomos se transformam rapidamente, mas naquela época eles não sabiam disso e acreditavam que apenas uma das estruturas deveria ser realizada. Somente o sinal de difração de elétrons não queria ser descrito por nenhuma dessas estruturas, e apenas uma combinação de sinais de ambas as conformações poderia explicar o padrão de difração observado (mais sobre isso pode ser encontrado no livro de I. Khargittai "Frank Science. Conversations with Famous Chemists").
O esquema do método em si é muito simples (veja a figura abaixo).
A coisa está acontecendo no vácuo.
- Elétrons rápidos são continuamente eliminados do cátodo, que são acelerados no campo anódico para energias de 40 a 60 keV.
- Elétrons suficientemente dispersos (mas rápidos) são focados por uma lente magnética, após o que se transformam em um feixe estreito.
- Uma câmara com uma substância é instalada perpendicularmente à viga. A amostra é aquecida até ferver e o vapor resultante entra em contato com o feixe de elétrons.
- Os elétrons são espalhados com sucesso pelas moléculas e voam silenciosamente mais longe, onde caem no filme.
- Geralmente na frente do filme colocar o chamado. dispositivo setorial. Esta é uma tela de rotação muito rápida, de forma incomum. O fato é que o elétron tem a probabilidade de se desviar de sua direção original (por um grande ângulo de dispersão theta ) cai muito rapidamente. Portanto, para amenizar essa diminuição de intensidade, o setor ofusca uniformemente a parte central do filme, deixando a parte mais aberta. O resultado é uma imagem mais uniformemente iluminada.
- A armadilha do feixe captura os elétrons que não estão dispersos (e existem muitos).
- Bem, para que as moléculas não voem por todo o dispositivo, sujando-o, elas são congeladas em uma armadilha fria resfriada por nitrogênio líquido.
O resultado é o mesmo padrão de difração de anéis concêntricos descrito pela equação de Debye (este é um sinal). Vários parâmetros moleculares podem ser extraídos diretamente dele.
Onde posso encontrar laboratórios de elétrons a gás?Ainda não existem muitos.
Mas na Rússia existem dois: Moscou (no Departamento de Química da Universidade Estadual de Moscou) e na Universidade Tecnológica Química de Ivanovo.
Espectroscopia de microondas
Esse método de estudo de moléculas é conhecido mais, por isso vou falar um pouco mais brevemente, usando a modificação mais moderna como exemplo: um espectrômetro de transformada de Fourier (como em russo, em suma, espectroscopia de microondas transformada por Fourier).
O design aqui já é mais complicado, pois requer um monte de eletrônicos diferentes (amplificadores, moduladores de frequência etc.). Vamos omitir tudo isso e falar apenas sobre o que está acontecendo dentro da câmara de vácuo.
- Um em frente ao outro, existem duas antenas de corneta (como a que abriu o estudo da relíquia ). Um deles serve como transmissor e o segundo é um receptor.
- Perpendicular a essas antenas é uma válvula que lança a amostra. Na maioria das vezes, é lançado na forma de vapor junto com um certo gás de arraste (geralmente gases inertes) no modo de expansão adiabática. Sob tais condições, as moléculas esfriam rapidamente a temperaturas próximas de 0 K, o que simplifica bastante o espectro, tornando-o mais suscetível à interpretação.
- Quando as moléculas enchem toda a câmara, a antena transmissora as irradia com um sinal modulado em frequência linear. Na representação de frequência, isso corresponde à soma de todas as frequências em um determinado intervalo.
- Algumas moléculas absorvem essa radiação em diferentes frequências transmitidas e entram em um estado excitado. Mas, depois de algum tempo, eles recuam, começando a irradiar o que captaram durante o impulso da antena transmissora. Este estol parece um sinal de oscilação decrescente ( decaimento de indução livre ). A segunda antena também a registra. Então, após a transformação de Fourier de gravar esse sinal no tempo, é obtido o espectro usual de frequência.
Ao contrário da difração de elétrons, que não era importante que tipo de molécula considerar, em uma espectroscopia de microondas uma molécula deve ter um momento dipolar constante (em casos raros, um momento dipolar magnético também é adequado, isso é típico para radicais, como uma molécula de O2). O sinal aqui é "intensidade de emissão vs. frequência ". As constantes rotacionais são extraídas desses espectros através de alguns modelos, dos quais a estrutura molecular é então extraída.
Bem-vindo ao zoológico de parâmetros moleculares!
Agora é hora de analisar quais parâmetros geométricos podemos obter de várias experiências. De fato, cada um dos tipos de quantidades indica que tipo de modelo foi usado para ajustar o sinal experimental (geralmente pelo método dos mínimos quadrados). A maioria desses parâmetros pode ser encontrada na revisão Kuchitsu K., Cyvin SJ // Em: Estrutura Molecular e Vibrações / Cyvin SJ (Ed.) - Amsterdã: Elsevier, 1972.- Ch.12. - P.183-211.
Vamos começar de novo com a eletronografia.
- rg= langler rangleT estrutura Este é apenas um conjunto de valores médios de distâncias interatômicas a uma determinada temperatura.
- ra= langler−1 rangle−1T esse valor é semelhante rg , mas é um pouco mais natural para descrever o padrão de difração.
- r alpha=rh,0=? . Esse valor não possui um significado físico claro e está totalmente vinculado ao modelo interpretativo. De fato, é o que se observa na cristalografia.
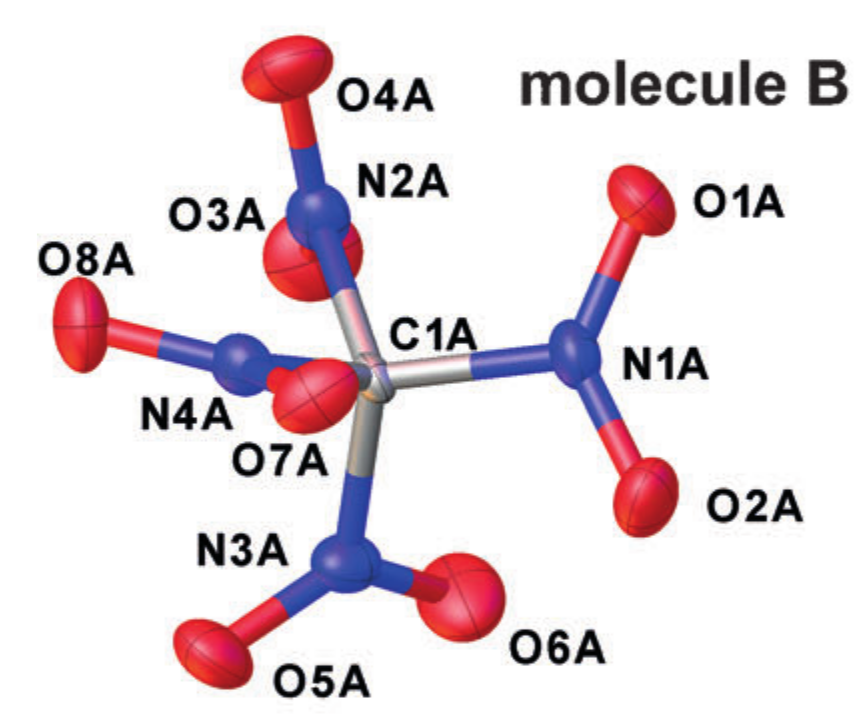
Exemplo cristalográfico r alpha estruturas (tetranitrometano no cristal). Adaptado de DOI: 10.1002 / ano.201704396
Cada átomo é aproximado por um elipsóide que descreve seu movimento vibracional, e as distâncias entre os centros das elipses resultantes são tomadas como distâncias interatômicas. Porém, essa simplificação da natureza do movimento dos átomos corresponde à introdução da aproximação do oscilador harmônico para oscilações e nem sempre funciona bem.
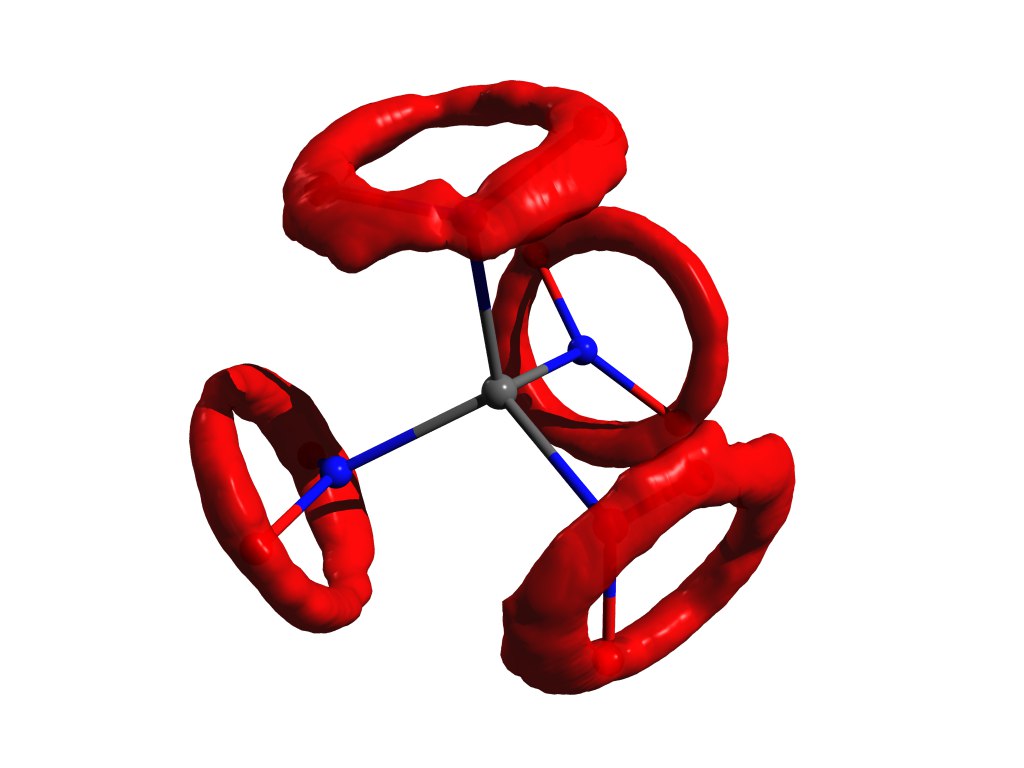
Um exemplo da distribuição de átomos quando a aproximação do oscilador harmônico não funciona. O mesmo tetranitrometano, mas no gás.
- rh1=??? . Essa baleia-peixe-
judô HEX não é descrita em poucas palavras, tem uma relação muito fraca com a realidade, mas possui propriedades maravilhosas: deve ser geometricamente consistente (veja abaixo) e pode ser facilmente calculada. Devido a isso, ela ganhou sua grande popularidade na comunidade eletronográfica.
Na espectroscopia de microondas, há um pouco menos de variantes estruturais.
- O mais fisicamente compreensível é rn= langlen| hatr|n rangle estrutura De fato, essa é a geometria média de um certo estado vibracional da molécula ( |n rangle ) Como geralmente trabalham com moléculas frias, geralmente observam uma estrutura específica dessa classe: r0 , ou seja, a geometria da molécula no estado vibracional do solo, quando os átomos produzem vibrações zero em torno de sua posição mais vantajosa.
- O mais popular é rs estrutura O subscrito "s" significa "substituição". Eles entendem da seguinte maneira: acredita-se que existem algumas coordenadas de átomos fixadas no espaço, depois fazem uma substituição isotópica de um átomo de um átomo na molécula e determinam a posição desse átomo alterando as constantes de rotação. A principal vantagem dessa tecnologia é a simplicidade. Menos: você só precisa de monossubstituição + nem todas as posições dos átomos podem ser estabelecidas assim + o significado físico desse modelo não é muito claro.
- Desenvolvimento lógico rs estruturas são r m -estruturas obtidas da montagem por mínimos quadrados ponderados em massa. Eles também precisam de moléculas isotopicamente substituídas, mas qualquer uma delas já é adequada.
E isso está longe de todos os tipos possíveis de estruturas ...
Mas o
grande simulador O usuário de algum pacote químico-quântico padrão (como o programa
Gaussian Evil Corporation ) ao usar um feitiço como "Opt" recebe o que é chamado de "geometria de equilíbrio", ou
re estrutura Essa é a configuração mais ideal dos núcleos, minimizando a energia eletrônica do sistema. E essas estruturas também podem ser extraídas da difração de elétrons e da espectroscopia rotacional, mas apenas para moléculas muito pequenas e simétricas e em combinação com outros métodos de pesquisa. Até agora, não deu certo.
E assim surge a pergunta: é correto comparar
re estrutura com alguns dos experimentais, olhando apenas para os erros experimentais?
A resposta aqui é simples:
não , é necessário estabelecer um erro adicional sobre possíveis diferenças sistemáticas. E um exemplo muito impressionante pode ser dado a isso: o efeito Bastiansen-Morino (consulte os artigos
DOI: 10.1107 / S0365110060002557 e
DOI: 10.1107 / S0365110060002545 ).
Suponha que tenhamos uma molécula do tipo CX
2 (isto é, CO
2 , CS
2 , etc.). Como deveríamos saber do curso de química da escola, essas moléculas têm uma estrutura linear (átomos de carbono e dois calcogênios X estão em uma linha reta).
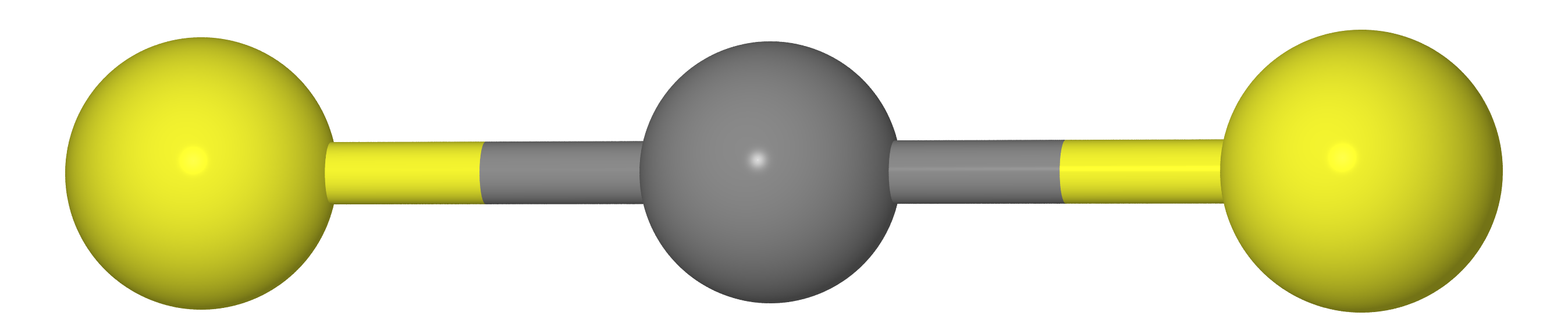
Isso significa que a distância entre os átomos X deve ser igual ao dobro do comprimento da ligação C - X (ou seja,
re( mathrmXX)=2re( mathrmCX) )

De qualquer forma, se medirmos as distâncias entre os átomos C e X (
rg( mathrmCX) ) e XX (
rg( mathrmXX) ) por difração de elétrons de gás, obtemos
rg( mathrmXX)<2rg( mathrmCX) , ou seja, a molécula acaba sendo curvada. A razão reside no fato de que a molécula faz o chamado
vibrações em tesoura , devido às quais os átomos X estão muito mais próximos um do outro do que na localização mais favorável (veja a figura abaixo).

De onde vem o efeito Bastansen-Morino. Imagem do artigo do
DOI: 10.1039 / C6CP05849C .
Portanto, se equipararmos a temperatura média
rg estrutura para o equilíbrio (
re ), concluiríamos erradamente que as moléculas de dióxido de carbono e dissulfeto de carbono são curvas.
É por isso que, ao comparar diferentes tipos de parâmetros geométricos, você deve sempre ter muito cuidado. Isso se aplica tanto à comparação de dados experimentais entre si quanto à comparação de experimentos e teorias.
Moléculas Modelo Padrão Moléculas
Agora vamos imaginar que desejamos de todo o coração simular o resultado de algum experimento com base em nosso modelo teórico para comparar a simulação com a realidade em uma batalha justa.
E aqui também é necessário ter cuidado, porque diferentes modelos de moléculas também têm seus limites de aplicabilidade. Vamos examinar isso usando o exemplo do Modelo Molecular Padrão.
Primeiro você precisa entender o que é o Modelo Padrão de Molécula. Os físicos do BAK têm seu próprio
modelo padrão , os astrônomos têm o
seu e os físicos têm seu próprio design básico, do qual mais tarde dançam. Porém, diferentemente dos modelos físicos, consideramos um conjunto de aproximações que permitem ao usuário obter o resultado de forma relativamente automática e rápida.
Para usuários gaussianosAgora lembramos o que está na base dos feitiços mágicos gaussianos "Opt" e "Freq".
O esquema geral das aproximações introduzidas é mais ou menos assim:
No fundo da qualidade está o nosso modelo padrão. Resuma brevemente todas as etapas de seu recebimento.
O modelo resultante é chamado RR-HO (@BO). Não tocaremos na aproximação de Born-Oppenheimer (BO), mas teremos que falar sobre o rotador rígido e o oscilador harmônico no quadro da química estrutural ...E o principal problema com essa aproximação é que a molécula não é rígida e suas vibrações são completamente harmônicas. Consequentemente, na realidade, precisamos da aproximação de um rotador não rígido e de um oscilador anarmônico. E a palavra-chave aqui é "anarmônica", ou seja, "Não é harmônico."Vamos falar sobre as moléculas mais simples: diatômica. Existem muitos exemplos deles: HCl, HBr, HI, CO, O 2 , N 2 , etc. etc.
Eles se distinguem de todas as moléculas pelo fato de terem apenas uma vibração: a extensão / compressão da distância interatômica.E essa é a distância entre os átomos que podemos medir na difração de elétrons gasosos (em uma variação da temperatura média,rg ) e na espectroscopia de rotação (média sobre, digamos, o estado vibracional do solo, ou seja, r0 )
E agora surge a principal questão do universo da vida e em geral:o que será rg e r0 na aproximação de um oscilador harmônico, e como isso se compara com a distância de equilíbrio re ?
Para uma resposta, você deve observar a superfície da energia potencial de uma molécula diatômica:
- , : . , .
- , , 2 :
- ( r→0 ), - , « » ,
- ( r→+∞ )
Como resultado, se a molécula diminui seu comprimento de ligação em relação à posição de equilíbrio, encosta-se à parede e, se aumenta, cai em um sofá macio. E a molécula não é tola, ficará mais no sofá do que batendo contra a parede. Portanto, a vibração média da distância será maior que o equilíbrio (re<r0,rg ), e isso é perceptível: esses deslocamentos são da ordem de 0,01 Å, que é maior que os erros de medição.
Portanto, mesmo se quisermos calcular algo mais parecido com um experimento, permanecendo dentro da estrutura do Modelo Padrão de Molécula (RR-HO @ BO), não teremos nada de novo; portanto, a própria geometria de equilíbrio participará da comparação.Conclusão do erro # 2
 Ilustração do artigo do DOI: 10.1002 / anie.201611308 .E a conclusão é terrivelmente simples e consiste em 2 partes.
Ilustração do artigo do DOI: 10.1002 / anie.201611308 .E a conclusão é terrivelmente simples e consiste em 2 partes.- Com uma comparação correta, todos os valores devem ter o mesmo significado.
- Se os valores forem diferentes, isso não deve ser esquecido.
Exemplos de erros em artigos científicos
"Obras hindus"
Na verdade, o principal local onde você pode encontrar isso são as revistas de baixo nível. Eles raramente contêm artigos com resultados interessantes, por isso foram escolhidos por Pesquisadores Líderes de países do segundo e mais Mundos (países do BRICS e seus seguidores menos bem-sucedidos). Por revistas de "baixo nível", aqui, entende-se não aqueles que publicam artigos como "O Rooter: Algoritmo para a Unificação Típica de Pontos de Acesso e Redundância", mas respeitam publicações científicas. No meu campo científico, as mais famosas "meias lavagens" são:(existem outros). Como você pode ver, de acordo com sinais formais, na ciência russa são consideradas publicações muito decentes. Mas chega uma onda de g ... conteúdo de qualidade questionável, que muito ainda está vazando.Como ilustração, peguei a última edição do Journal of Molecular Structure e examinei o índice, e pronto:S. Sathiya, M. Senthilkumar, C. Ramachandra Raja, crescimento de cristal, análise de superfície de Hirshfeld, estudo DFT e estudos NLO de terceira ordem de tioureia 4 dimetil aminobenzaldeído // J. Mol. Struct., V. 1180 (2019), PP. 81-88.
https://doi.org/10.1016/j.molstruc.2018.11.067
A estrutura geral desse trabalho é muito despretensiosa.- «» ( ) . .
- (), . — , ( , ) , . , - Gaussian, « ». … 1 2 , .. - .
- , /Vis, .
- Em visualizadores moleculares padrão, como GaussView (grunhido), belas imagens são desenhadas, mas sempre com baixa qualidade.
- Não são tiradas conclusões substantivas: “experimentamos muito, contamos muito, trouxemos tabelas e quadros ⇒ nós somos ótimos, nos dê doces. ”
Mas dos meus favoritos: artigoM. Govindarajan, M. Karabacak, FT-IR, FT-Raman e investigação espectral UV; análise de estimação de frequência computada e cálculos de estrutura eletrônica em 1-nitronaftaleno // Spectrochimica Acta Parte A: Espectroscopia Molecular e Biomolecular, V. 85 (2012), PP. 251-260,
https://doi.org/10.1016/j.saa.2011.10.002.
Nele, o espectro na fase sólida foi estupidamente registrado e, em seguida, interpretado com base em cálculos medíocres no modelo HO na fase gasosa. Mas o truque é que eles nem conseguiam fazer cálculos normais, o
que eles educadamente sugeriram no comentário ao artigo .
No entanto, o termo "obras hindus" (como o termo "
código hindu ") refere-se longe de apenas obras provenientes do subcontinente misterioso correspondente.
Se você for ao maravilhoso local de
Cyberleninka , então
você olha para o Abismo, e o Abismo olha para você, você pode descobrir muitas coisas interessantes. Ao procurar por "químico quântico" (com / sem a condição adicional "rsa"), foi possível encontrar muitas coisas úteis. Como uma grande quantidade de trabalhos "químico-quânticos" foi dedicada ao estudo de cavalos esféricos no vácuo (isto é, cálculos sem referência à realidade), eles não estavam relacionados a este texto. Mas entre eles, essas três obras foram perdidas:
Fiquei especialmente satisfeito com o último, porque a "avaliação da adequação" era uma comparação inadequada de estruturas em diferentes fases (gás x cristal) e com diferentes significados (
re vs.
r alpha ) - esta é realmente a altura da adequação.
Isso acontece em boas revistas?
Sim, existem "erros" lá.
Não tenha medo, tudo acabou bem!Felizmente, na história a que agora nos voltamos, uma atitude descuidada em relação aos valores experimentais / teóricos não levou a consequências graves e terríveis. E o trabalho, apesar da omissão real, não perde a frieza e o significado.
Além disso, no final tudo terminou em geral super: a publicação de um artigo impressionante, onde a justiça triunfou.
Estamos falando de uma das moléculas orgânicas com uma ligação C - C única e extremamente longa:
1,1'-bisdiadamantano :

Por que essa molécula é legalSe olharmos para
os livros de química da escola ou da
universidade em química, no
Wiki (ou
até mesmo no Google ), descobriremos que o comprimento padrão de uma única ligação C - C é 1,54 Å.
Assim, o comprimento experimental da ligação simples central em 1,1'-bisdiadamantano
re=1.630 pm0.005 Å, quase 0,08 Å a mais (isso é dofiga)!
Essa extensão de uma única ligação C - C ocorre porque as duas peças em forma de diamante que essa conexão mantém são muito grandes, portanto se repelem. Mas, como lembramos do exemplo do Ferroceno, também temos atração devido às forças de dispersão (Londres). E devido ao tamanho doentio dessas metades da molécula, há uma grande atração de dispersão entre as peças. Ele não permite que essa ligação central se rompa; portanto, essa molécula pode (relativamente facilmente) ser vaporizada sem quebrá-la ao longo do caminho.
A conexão unária central C - C é muito longa e, portanto, foi muito interessante comparar como os métodos teóricos reproduzem a realidade. E a realidade está nos primeiros artigos, aqui estão eles:
Puta merda!O primeiro artigo está na Nature, a revista mais legal, e o segundo na JACS, a mais respeitada revista puramente química !!! Poucas pessoas sonharam com isso, mas essa colaboração científica entre a Alemanha e a Ucrânia é muito legal.
foi representado apenas por dados cristalográficos. Como resultado, comparando coisas difíceis comparáveis entre si, sem fazer as
devidas correções para a diferença de parâmetros, eles finalmente chegaram à conclusão de que o comprimento da ligação C - C central no gás é de 1,655 Å, ultrapassado em 0,02 Å. E isso é significativamente mais do que o erro experimental.
Felizmente, no final, eles colaboraram com especialistas nessas questões
e ,
no final, receberam a resposta correta (um
pequeno resumo popular deste trabalho também pode ser encontrado no N + 1 ).
Você precisa de uma comparação?

Depois de tudo o que escrevi sobre a exatidão das comparações, pode surgir uma pergunta razoável: é então necessário comparar os resultados dos cálculos e os resultados das experiências entre si?
É necessário! Assim como você precisa!Existe uma declaração famosa (da qual não consegui encontrar um autor confiável):
Ninguém acredita em cálculos teóricos, exceto aquele que os fez.
Todo mundo acredita em resultados experimentais, exceto aquele que os obteve.
Traduzido para o russo, parece: ninguém acredita nos cálculos teóricos, exceto aquele que os fez, mas todo mundo acredita nos dados experimentais, exceto aquele que os recebeu.
Mas na ciência é necessário que todos acreditem (ou a maioria), e o experimento é a única medida, pois relaciona o que calculamos à Realidade.
Há um ensaio maravilhoso (acesso aberto) no segundo periódico químico mais importante sobre o que um pesquisador ou uma pessoa comum que lê artigos científicos e / ou notícias deve fazer:
Mata R., Suhm M. // Angew. Chem. Int. Ed., 56 (2017), DOI: 10.1002 / ano.201611308(a propósito, eu já dei um link para ele, já que uma foto, de Ricardo Mata, é deste artigo).
As conclusões deste ensaio fornecem recomendações aos teóricos e experimentadores da simulação. Vou dar a eles aqui (em tradução e uma pequena revisão) como uma palavra final para este post.
- O teórico deve:
- forneça não apenas métodos e tentativas bem-sucedidas, mas também descreva as falhas dos métodos (especialmente se esses métodos forem populares),
- descreva sua metodologia bem e completamente,
- onde é possível fornecer estimativas (ou descrição) de erros e aproximações e simplificações importantes aceitas.
- Os experimentadores, por sua vez, devem:
- cutucar os teóricos com seus dados experimentais, que eles poderiam usar como benchmarks (padrões),
- mostrar à comunidade científica dados experimentais incompreensíveis, onde a teoria (ou experimentos adicionais) ajudaria na explicação,
- falar em
território estrangeiro de conferências teóricas com seus dados,
história pessoalA propósito, acabei de conhecer o segundo autor deste artigo, que é um experimentador, em um simpósio teórico.
- retire de seus dados experimentais coisas que sejam o mais acessível possível para comparação com a teoria,
Todas as melhores e corretas comparações! E lembre-se: somente quem não faz nada não está enganado.
PS
Como um posfácio, eu gostaria de fornecer uma pequena lista de bancos de dados onde você pode desenterrar vários dados experimentais de moléculas.
Bancos de dados estruturais
Latas cristalográficas
A maneira mais fácil de determinar a estrutura de uma molécula em um cristal é porque PCA é uma rotina. Portanto, se você não sabe como é a molécula, vá para os bancos de dados cristalográficos (locais onde são coletadas quase todas as estruturas de substâncias que já foram amontoadas em um goniômetro e iluminadas por um feixe de partículas de ondas curtas). Como existem muitos desses bancos, darei apenas os mais famosos (uma lista mais completa pode ser encontrada no
Wiki ).
- Banco de Dados de Estrutura Cristalina Inorgânica ( ICSD ). Não se trata inteiramente de moléculas, é possível encontrar principalmente estruturas de diferentes sais, metais, cerâmicas etc. Esta base é suportada pela Universidade de Tecnologia Karlsruhev, portanto, o acesso a ela é pago e não é barato. Mas se isso, o site dela .
- Banco de Dados Estruturais de Cambridge ( CSD ). Talvez a maior base cristalográfica do mundo. Quase um milhão de estruturas! Estes são principalmente cristais moleculares orgânicos e organometálicos. E esse banco de dados é grátis! Para adicionar. uma taxa, é claro, você pode obter uma opção interessante, como uma pesquisa na estrutura desenhada da molécula, mas isso já é excedente. O site .
- Banco de Dados Aberto de Cristalografia ( COD ). Bem, também algum tipo de banco de dados. O que há, eu realmente não sei, mas pelo menos ele existe. O site .
- Bem, para aqueles que estão interessados em biologia, existe um Protein Data Bank ( PDB ). Um banco de dados aberto, onde você pode baixar as estruturas de proteínas enormes e assustadoras (mas em cristais). O site .
A estrutura das moléculas em um gás
Aqui as coisas são um pouco piores, porque experimentos para estudar a estrutura das moléculas livres são muito mais complicados, tanto para a realização (pelo menos é necessário um alto vácuo) quanto para a interpretação.
Portanto, existem substancialmente menos bancos de dados.
- O maior banco de dados para moléculas livres é a DOCumentation MOlecular GAsphase (ou MOGADOC ). Baseia-se na Universidade de Ulm e é um investimento muito caro. Mas, se houver, o site está aqui .
- Se você deseja conhecer as estruturas experimentais de equilíbrio de moléculas de 100%, essa é a base de dados de comparação e comparação de química computacional do NIST ( CCCBDB ). Quase tudo puramente experimental re -estruturas podem ser encontradas lá, mas existem outros nishtyaks suficientes lá. O site .
- O último bastião é o projeto subdesenvolvido do MolWiki . Mas não há muitas estruturas que podem ser encontradas lá, apenas aquelas que foram recebidas na Universidade de Bielefeld, na Universidade Estadual de Moscou e no ISTU nos últimos anos (e mesmo assim nem todas). O site .
Onde encontrar as características espectrais das moléculas?
 Retirado de xkcd.com
Retirado de xkcd.comExistem muito mais bancos de dados aqui, pois remover o espectro sem interpretação é uma tarefa muito mais simples do que obter uma estrutura (não há necessidade de construir modelos e provar que eles estão corretos). Além disso, os espectros são de grande valor aplicado: eles podem ser usados para determinar a composição das amostras, seja água de um rio vizinho ou um sinal de uma nuvem molecular (ou mesmo de uma atmosfera de exoplaneta, veja a ilustração acima).
Sim, a propósito, todos os links nesta seção serão para liberar bancos de dados.
- Webbook de Química do NIST . Talvez o melhor livro do mundo. Neste banco de dados, você pode encontrar espectros de massa de IV, UV / Vis, para um monte de moléculas. E também, parâmetros termoquímicos e, às vezes, cinéticos! Em geral, se um químico não sabe sobre a existência desse site, ele não é um químico. O site .
- Banco de dados de absorção molecular de transmissão de alta resolução ( HITRAN ). O projeto do Centro de Astrofísica Harvard-Smithsonian, do qual é óbvio que os espectros mostrados lá são usados para identificar moléculas em espaços interestelares e circunstanciais (por exemplo, assim ). O site .
- Semelhante ao anterior, o projeto The Cologne Database for Molecular Spectroscopy, da parte astrofísica da Universidade de Colônia. O site .
- Bem, como último exemplo, darei o projeto ExoMol . Estes não são espectros experimentais inteiramente puros, mas este é um excelente exemplo da interação entre teoria e experimento: com base em dados experimentais de alta precisão e em cálculos de nível muito alto, os espectros de moléculas simples são previstos sob condições diferentes (inclusive extremas). A ênfase principal aqui é nos biomarcadores; portanto, quando os astrônomos veem os espectros dos exoplanetas, eles podem facilmente identificar as moléculas que já conhecemos neles. O site .
PPS
Se houver erros / algo permanecer incompreensível, escreva nos comentários - vou corrigir / tentar explicar melhor.