Não faz muito tempo, li o romance de ficção científica
"O Problema dos Três Corpos", de Liu Qixin . Nele, alguns alienígenas tinham um problema - eles não sabiam como, com precisão suficiente, calcular a trajetória de seu planeta natal. Ao contrário de nós, eles viviam em um sistema de três estrelas, e o “clima” no planeta dependia fortemente de sua localização relativa - do calor incinerador ao frio gélido. E decidi verificar se podemos resolver esses problemas.
Física do fenômeno
Para entender o problema, é necessário lidar com a física do fenômeno. No quadro da teoria clássica, a força atrativa de dois corpos é determinada pela lei
de Newton :
v e c F ( v e c r 1 , v e c r 2 ) = - G m 1 m 2 f r a c v e c r 1 - v e c r 2 | v e c r 1 - v e c r 2 | 3 ,
onde
vecr1, vecr2 - a posição dos corpos no espaço,
m1,m2 - massas de corpos,
G - constante gravitacional.
No sistema de
N Os corpos de cada um deles serão afetados pela força de atração do resto, que é expressa pela equação:
vecFn=−G sumk neqnmnmk frac vecrn− vecrk| vecrn− vecrk|3.
Usando
a segunda lei de Newton, escrevemos a aceleração para cada partícula:
vecan= vecFn/mn=−G sumk neqnmk frac vecrn− vecrk| vecrn− vecrk|3.
Lembrando que a aceleração é a segunda derivada da coordenada, obtemos uma equação diferencial parcial de segunda ordem, que deve ser resolvida para obter a trajetória de cada corpo:
\ frac {\ parcial ^ 2 \ v {{}} n \ {parcial t ^ 2} = f_n = -G \ sum_ {k \ neq n} m_k \ frac {\ vec {r} _n - \ vec {r } _k} {| \ vec {r} _n - \ vec {r} _k | ^ 3}.
É importante notar aqui que a complexidade de calcular uma função
fn é igual a
O(N2) e aumenta significativamente com o aumento do número de corpos em interação.
Matemática
O primeiro e mais simples método para resolver equações diferenciais é
o método de Euler , projetado para resolver equações da forma:
fracdydx=f(x,y).
Após a transição para a região discreta, obtemos:
yi=yi−1+hf(xi−1,yi−1), quadi=1,2,3, dots,m,
onde
h A etapa de integração e
m - o número de etapas de integração. Assim, se precisarmos calcular a posição dos corpos de cada vez
T então devemos fazer
m=T/h etapas de integração. Aqui o primeiro problema é visível - se
T grande, precisamos dar um grande número de etapas de integração.
Para aplicar o método Euler ao nosso problema, ele deve ser reduzido a um sistema de primeira ordem. Para fazer isso, introduzimos uma variável adicional - velocidade das partículas:
frac parcial vvn parcialt=fn, frac parcial vecrn parcialt= vecvn.
O segundo problema na solução de sistemas de equações diferenciais é a precisão da solução e seu controle. A precisão pode ser aprimorada de duas maneiras: diminuindo a etapa de integração e escolhendo um método com uma ordem de precisão mais alta. Ambos os métodos levam ao aumento da complexidade computacional, mas de maneiras diferentes. Por exemplo, você pode usar o
método Runge-Kutta clássico de
quarta ordem ; ele requer quatro cálculos de função
fn a cada passo, mas tem uma ordem de precisão
O(h4) (para comparação, o método de Euler tem uma ordem de precisão
O(h) e requer um cálculo
fn ) A precisão da solução também pode ser controlada de várias maneiras: compare com a solução analítica, resolva usando métodos diferentes ou com etapas diferentes e compare os resultados, controle parâmetros e limitações de terceiros que a solução deve cumprir.
Além disso, cada um desses métodos tem suas desvantagens. As decisões analíticas podem estar ausentes ou, mesmo na maioria dos casos, completamente ausentes. Por exemplo, para nossa tarefa
N solução analítica tel é apenas para
N=2 , mas mesmo isso é suficiente para testar a precisão dos métodos. A resolução de um problema por dois métodos ou com etapas diferentes aumenta o tempo de computação, mas essa abordagem pode ser aplicada a quase qualquer tarefa. Nem todas as tarefas têm limitações, mas as nossas têm: em cada etapa da integração, podemos controlar a implementação
das leis de conservação . Essa abordagem também aumenta a complexidade do cálculo, mas há muito por onde escolher; o cálculo da soma do momento ou do momento angular de todas as partículas tem complexidade da ordem
O(N) , enquanto o cálculo da energia total de um sistema tem complexidade de pedidos
O(N2)Nota sobre o cálculo da energia totalNo nosso caso, a energia total do sistema consiste em duas partes - energia cinética e potencial. A energia cinética consiste na soma das energias cinéticas de todos os corpos. Para calcular a energia potencial, precisamos adicionar as energias potenciais de cada partícula no campo gravitacional das partículas restantes, portanto, precisamos adicionar
O ( N 2 ) termos. A dificuldade é que todos os termos são de uma ordem muito diferente e, mesmo com cálculos de precisão dupla, não é possível calcular esse valor com uma precisão suficiente para comparação em etapas diferentes. Para superar esse problema, foi necessário aplicar a soma de acordo com
o algoritmo Kahan .
Fig 1. Um exemplo de uma trajetória elíptica.
Considere o caso simples de um satélite se movendo em uma órbita elíptica ao redor da Terra. À medida que o satélite se aproxima da Terra, sua velocidade aumenta e, ao se afastar da Terra, diminui, consequentemente, a possibilidade de diminuir o passo de integração no tempo na parte verde da órbita e aumentá-lo em vermelho e azul sem alterar a precisão da solução. Vamos tentar comparar com mais detalhes.
Tabela 1. Métodos investigados para resolver equações diferenciaisPara selecionar o melhor método para nossa tarefa, compararemos vários métodos conhecidos. Para isso, simulamos a colisão de dois sistemas de corpos
N=512 e medir a mudança relativa na
energia total ,
momento e seu
momento no final da simulação (tempo máximo de simulação
Tmax=2,5 ) Nesse caso, variaremos a etapa e os parâmetros dos métodos de integração e mediremos o número de chamadas de função
fn , respectivamente, aqueles métodos que com um número menor de chamadas levarão a menos perdas, consideraremos mais aceitáveis.

| 
|
| a) | b) |
| Figura 2. Mudança relativa na energia a), momento b), no final da simulação do sistema N=512 corpos por vários métodos, dependendo do número de cálculos de funções fn precisão dupla |
A partir dos gráficos da Figura 2, é possível observar que a melhor proporção da quantidade de cálculo da função
fn e a mudança relativa na energia do sistema dos corpos nos métodos de Adams e Dorman-Prince de quinta ordem. Também é visto que para todos os métodos com um aumento no número de cálculos
fn a mudança relativa no momento do sistema aumenta. Para uma mudança relativa na energia, isso também é perceptível, mas apenas para alguns métodos que podem atingir o limite
dE/E0<10−12 . Esse efeito não pode mais ser atribuído ao erro dos métodos, mas ao erro dos cálculos, e um aumento adicional na precisão é possível apenas junto com um aumento na precisão dos cálculos com ponto flutuante.

| 
|
| a) | b) |
| Fig. 3. Mudança relativa na energia a), momento b), no final da simulação do sistema N=512 corpos por vários métodos, dependendo do número de cálculos de funções fn com precisão quádrupla (__float128) |
As Figuras 3a e 3b mostram que o uso de cálculos com precisão quádrupla pode reduzir as perdas de energia relativas em até
10−23 , mas você precisa entender que o tempo de computação é aumentado em duas ordens de magnitude em comparação com a precisão dupla. Como no caso de cálculos de dupla precisão, a melhor razão de precisão e o número de cálculos de funções
fn possuem métodos de Adams de quinta ordem e Dorman-Prince.
Os
métodos Dorman-Prince e Werner pertencem à classe de
métodos aninhados e podem calcular simultaneamente duas soluções com alta e baixa ordem de precisão (para o método Dorman-Prince, ordens 5 e 4, e para o método Werner, ordens 6 e 5). Se essas duas soluções forem muito diferentes, poderemos dividir a etapa atual de integração em outras menores. Isso nos permite alterar dinamicamente a etapa de integração e reduzi-la apenas nas áreas em que é necessária.
Vamos comparar os métodos de quinta ordem Dorman-Prince, Werner e Adams com mais detalhes, durante um intervalo de simulação mais longo do nosso sistema (
Tmax=300 )

|
a) A mudança relativa de energia, momento e seu momento no processo de modelagem
|

|
b) Número de cálculos de funções fn no intervalo de simulação DeltaT=0,3
|
Fig. 4. Dependências da mudança relativa nos parâmetros físicos do sistema e o número de cálculos de funções fn Modelagem periódica com o método de etapas variáveis de Dorman-Prince h=10−5 pontos10−9
|

|
a) A mudança relativa de energia, momento e seu momento no processo de modelagem
|

|
b) Número de cálculos de funções fn no intervalo de simulação DeltaT=0,3
|
Fig. 5. Dependências da mudança relativa nos parâmetros físicos do sistema e o número de cálculos de funções fn modelagem versus tempo pelo método Werner com uma etapa variável h=10−5 pontos10−9
|

|
Fig. 6. Mudança relativa de energia, momento e seu momento no processo de modelagem pelo método de Adams-Bashfort de quinta ordem com um passo h=10−6
|

|
Fig 7. Dependências do número de cálculos de funções fn para métodos Adams, Dorman-Prince e Werner de quinta ordem a partir do tempo de simulação
|
Pode-se ver que no estágio inicial da evolução do nosso sistema (
T<25 ) os três métodos mostram características semelhantes, mas em estágios posteriores alguns eventos ocorrem no sistema, como resultado dos erros nos principais parâmetros do sistema (energia total, momento e seu momento) saltam acentuadamente. Mas os métodos Dorman-Prince e Werner lidam com essas mudanças muito melhor devido à capacidade de reduzir a etapa de integração em seções "complexas", como resultado do qual o número de cálculos de funções aumenta
fn como visto nas figuras 4b e 5b, mas o número total de cálculos
fn métodos aninhados são menores que o método Adams-Bashfort, como pode ser visto na Figura 7.
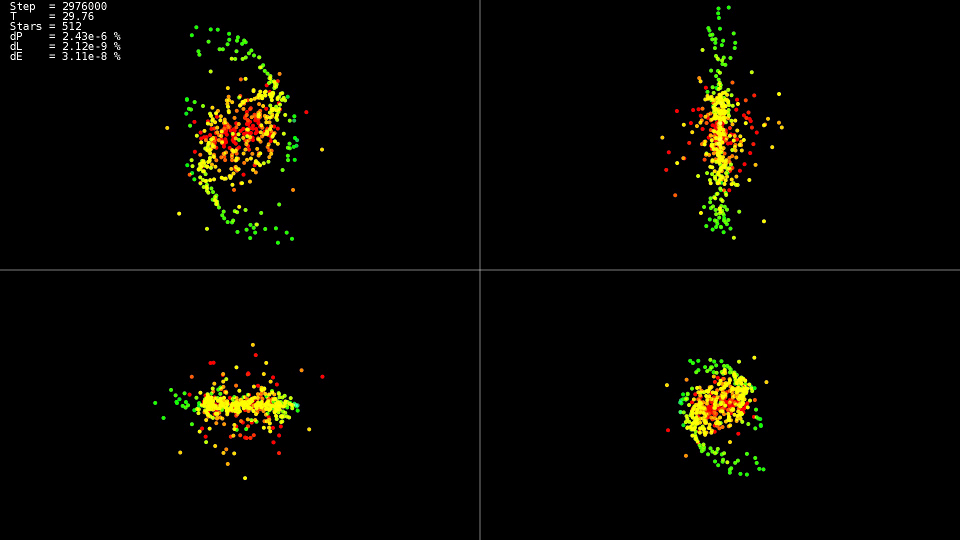
Eu me pergunto o que aconteceu com o sistema nesses momentos
|
Vídeo 1. Modelando um sistema de 512 corpos. Método Dorman-Prince. Etapa dinâmica h = 10 - 5 p o n t o s 10 - 9
|
O vídeo demonstra que até o momento
T=25 o movimento é relativamente calmo e após uma colisão dos centros das “galáxias”, o que leva a uma mudança acentuada nas trajetórias e um forte aumento nas velocidades de algumas partículas. Além disso, para manter a precisão da solução, é necessário reduzir a etapa de integração. Os métodos aninhados podem fazer isso automaticamente; os gráficos mostram que em algumas partes da evolução do sistema, a etapa de integração foi reduzida em quase duas ordens de magnitude com
10−5 antes
h=10−7 . Ao usar o método Adams e essa etapa em todo o intervalo da evolução do sistema, não teríamos esperado por uma solução.
Sumário
Para a solução, é melhor usar métodos aninhados que permitem controlar dinamicamente a etapa de integração e reduzi-la apenas em seções "complexas" da trajetória.
Não siga os métodos de ordem mais alta. Mesmo ao usar o tipo de dados 'double', eles não atingem seus recursos potenciais, usar os tipos de dados com maior precisão aumenta muito o tempo necessário para resolver o problema.
Implementação de CPU
Agora que a escolha de um método para resolver equações está definida, vamos tentar descobrir o cálculo da força de interação para cada partícula. Temos um ciclo duplo para todas as partículas:
Código de implementação 'simples'for(size_t body1 = 0; body1 < count; ++body1) { const nbvertex_t v1(rx[ body1 ], ry[ body1 ], rz[ body1 ]); nbvertex_t total_force; for(size_t body2 = 0; body2 != count; ++body2) { if(body1 == body2) { continue; } const nbvertex_t v2(rx[ body2 ], ry[ body2 ], rz[ body2 ]); const nbvertex_t force(m_data->force(v1, v2, mass[body1], mass[body2])); total_force += force; } frx[body1] = vx[body1]; fry[body1] = vy[body1]; frz[body1] = vz[body1]; fvx[body1] = total_force.x / mass[body1]; fvy[body1] = total_force.y / mass[body1]; fvz[body1] = total_force.z / mass[body1]; }
As forças gravitacionais de cada corpo são calculadas independentemente e, para usar todos os núcleos do processador, basta escrever a diretiva OpenMP antes do primeiro ciclo:
Um pedaço de código da implementação do 'openmp' #pragma omp parallel for for(size_t body1 = 0; body1 < count; ++body1)
Porque Como cada corpo interage com cada um deles, para reduzir o número de interações do processador com a RAM e melhorar o uso do cache, temos a capacidade de carregar parte dos dados no cache e usá-lo repetidamente:
Código de implementação 'openmp + block' #pragma omp parallel for for(size_t n1 = 0; n1 < count; n1 += BLOCK_SIZE) { nbcoord_t x1[BLOCK_SIZE]; nbcoord_t y1[BLOCK_SIZE]; nbcoord_t z1[BLOCK_SIZE]; nbcoord_t m1[BLOCK_SIZE]; nbvertex_t total_force[BLOCK_SIZE]; for(size_t b1 = 0; b1 != BLOCK_SIZE; ++b1) { size_t local_n1 = b1 + n1; x1[b1] = rx[local_n1]; y1[b1] = ry[local_n1]; z1[b1] = rz[local_n1]; m1[b1] = mass[local_n1]; } for(size_t n2 = 0; n2 < count; n2 += BLOCK_SIZE) { nbcoord_t x2[BLOCK_SIZE]; nbcoord_t y2[BLOCK_SIZE]; nbcoord_t z2[BLOCK_SIZE]; nbcoord_t m2[BLOCK_SIZE]; for(size_t b2 = 0; b2 != BLOCK_SIZE; ++b2) { size_t local_n2 = b2 + n2; x2[b2] = rx[local_n2]; y2[b2] = ry[local_n2]; z2[b2] = rz[local_n2]; m2[b2] = mass[n2 + b2]; } for(size_t b1 = 0; b1 != BLOCK_SIZE; ++b1) { const nbvertex_t v1(x1[ b1 ], y1[ b1 ], z1[ b1 ]); for(size_t b2 = 0; b2 != BLOCK_SIZE; ++b2) { const nbvertex_t v2(x2[ b2 ], y2[ b2 ], z2[ b2 ]); const nbvertex_t force(m_data->force(v1, v2, m1[b1], m2[b2])); total_force[b1] += force; } } } for(size_t b1 = 0; b1 != BLOCK_SIZE; ++b1) { size_t local_n1 = b1 + n1; frx[local_n1] = vx[local_n1]; fry[local_n1] = vy[local_n1]; frz[local_n1] = vz[local_n1]; fvx[local_n1] = total_force[b1].x / m1[b1]; fvy[local_n1] = total_force[b1].y / m1[b1]; fvz[local_n1] = total_force[b1].z / m1[b1]; } }
Uma otimização adicional consiste em retirar o conteúdo da função de calcular a força no ciclo principal e eliminar a divisão e multiplicação pela massa corporal m1 [b1]. Além do fato de termos reduzido um pouco os cálculos, o compilador poderá aplicar instruções vetoriais do processador SSE e AVX em um ciclo tão expandido.
Código de implementação 'openmp + block + optimization' #pragma omp parallel for for(size_t n1 = 0; n1 < count; n1 += BLOCK_SIZE) { nbcoord_t x1[BLOCK_SIZE]; nbcoord_t y1[BLOCK_SIZE]; nbcoord_t z1[BLOCK_SIZE]; nbcoord_t total_force_x[BLOCK_SIZE]; nbcoord_t total_force_y[BLOCK_SIZE]; nbcoord_t total_force_z[BLOCK_SIZE]; for(size_t b1 = 0; b1 != BLOCK_SIZE; ++b1) { size_t local_n1 = b1 + n1; x1[b1] = rx[local_n1]; y1[b1] = ry[local_n1]; z1[b1] = rz[local_n1]; total_force_x[b1] = 0; total_force_y[b1] = 0; total_force_z[b1] = 0; } for(size_t n2 = 0; n2 < count; n2 += BLOCK_SIZE) { nbcoord_t x2[BLOCK_SIZE]; nbcoord_t y2[BLOCK_SIZE]; nbcoord_t z2[BLOCK_SIZE]; nbcoord_t m2[BLOCK_SIZE]; for(size_t b2 = 0; b2 != BLOCK_SIZE; ++b2) { size_t local_n2 = b2 + n2; x2[b2] = rx[local_n2]; y2[b2] = ry[local_n2]; z2[b2] = rz[local_n2]; m2[b2] = mass[n2 + b2]; } for(size_t b1 = 0; b1 != BLOCK_SIZE; ++b1) { for(size_t b2 = 0; b2 != BLOCK_SIZE; ++b2) { nbcoord_t dx = x1[b1] - x2[b2]; nbcoord_t dy = y1[b1] - y2[b2]; nbcoord_t dz = z1[b1] - z2[b2]; nbcoord_t r2(dx * dx + dy * dy + dz * dz); if(r2 < NBODY_MIN_R) { r2 = NBODY_MIN_R; } nbcoord_t r = sqrt(r2); nbcoord_t coeff = (m2[b2]) / (r * r2); dx *= coeff; dy *= coeff; dz *= coeff; total_force_x[b1] -= dx; total_force_y[b1] -= dy; total_force_z[b1] -= dz; } } } for(size_t b1 = 0; b1 != BLOCK_SIZE; ++b1) { size_t local_n1 = b1 + n1; frx[local_n1] = vx[local_n1]; fry[local_n1] = vy[local_n1]; frz[local_n1] = vz[local_n1]; fvx[local_n1] = total_force_x[b1]; fvy[local_n1] = total_force_y[b1]; fvz[local_n1] = total_force_z[b1]; } }
Tabela 2. Dependência do tempo de cálculo (em segundos) da função fn no número de corpos interagindo N para várias implementações de CPU| N | 2048 | 4096 | 8192 | 16384 | 32768 |
|---|
| simples | 0,0425 | 0,1651 | 0,6594 | 2,65 | 10,52 |
| openmp | 0,0078 | 0,0260 | 0,1079 | 0,417 | 1.655 |
| openmp + block + otimização | 0,0037 | 0,0128 | 0,0495 | 0,194 | 0,774 |
Parâmetros do sistema:- sistema: Debian 9, Intel Core i7-5820K (6 núcleos)
- compilador: gcc 6.3.0
É claramente visto que a versão com suporte ao OpenMP é acelerada em seis vezes, exatamente em termos de número de núcleos, e a versão otimizada é mais rápida em pouco mais de duas vezes. Portanto, com a otimização, você não deve confiar apenas no paralelismo. Curiosamente, nos cálculos de fluxo único, o processador trabalhava com uma frequência de 3,6 GHz, na versão paralela (openmp) reduzia a frequência para 3,4 GHz e, no paralelo e otimizado (openmp + bloco + otimização), reduzia para 3,3 GHz, mas isso isso não a impediu de trabalhar 13,6 vezes mais rápido. Também é visto que um aumento no tempo de computação com um aumento no tamanho do problema é quadrático e um aumento adicional
N torna a tarefa insolúvel em um tempo razoável.
Implementação de GPU
Mas há um desejo de fazer cálculos ainda mais rápidos. Existem várias instruções disponíveis para aceleração: computação em GPU, aproximação de funções
fn . Primeiro, para a computação da GPU, escolhi a tecnologia OpenCL. Para um trabalho mais conveniente, a biblioteca
CLHPP foi usada. A principal vantagem do OpenCL é que o código pode ser executado no processador e na GPU, o que simplifica a gravação e a depuração, além de expandir a lista de hardware a ser executada. A ferramenta
Oclgrind ajuda na depuração, que em tempo de execução mostra chamadas incorretas da API OpenCL e problemas de acesso à memória.
Opencl
Para começar a usar o OpenCL, você precisa obter uma lista de plataformas disponíveis. As plataformas mais comuns são AMD, Intel e NVidia.
Código std::vector<cl::Platform> platforms; cl::Platform::get(&platforms);
Em seguida, depois de escolher uma plataforma, você precisa selecionar o dispositivo de computação que essa plataforma representa:
Código const cl::Platform& platform(platforms[platform_n]); std::vector<cl::Device> all_devices; platform.getDevices(CL_DEVICE_TYPE_ALL, &all_devices);
E no final da fase preparatória, é necessário criar um contexto e filas dentro das quais a memória será alocada e os cálculos serão realizados. Por exemplo, um contexto que combina todos os dispositivos de computação de uma plataforma selecionada é criado da seguinte maneira:
Código de criação de contexto e fila cl::Context context(all_devices); std::vector<cl::CommandQueue> queues; for(cl::Device device: all_devices) queues.push_back(cl::CommandQueue(context, device));
Para baixar o código-fonte para um dispositivo de computação, ele deve ser compilado, a classe cl :: Program é destinada a isso.
Código de compilação do kernel std::vector< std::string > source_data; cl::Program::Sources sources; for(int i = 0; i != files.size(); ++i) { source_data.push_back(load_program(files[i]));
Para descrever os parâmetros de uma função (kernel) executada em um dispositivo de computação, existe um modelo de cl: make_kernel.
Exemplo de kernel de cálculo de força de interação typedef cl::make_kernel< cl_int, cl_int,
Então tudo é simples: declaramos uma variável com o tipo de kernel, passamos o programa compilado e o nome do kernel computacional para ele, podemos iniciar o kernel quase como uma função normal.
Código de Lançamento do Kernel ComputeBlock fcompute(prog, "ComputeBlockLocal"); cl::NDRange global_range(device_data_size); cl::NDRange local_range(block_size); cl::EnqueueArgs eargs(ctx.m_queue, global_range, local_range); fcompute(eargs, ... );
O núcleo computacional do OpenCL é muito semelhante à opção 'openmp + block + optimization' da CPU, diferentemente da versão da CPU, o primeiro ciclo é controlado usando o OpenCL (o intervalo do ciclo é determinado pela variável global_range do código de inicialização do kernel, e o número da iteração atual está disponível no kernel usando a função get_global_id (0)). Primeiro, parte dos dados do corpo é carregada da memória local e processada. A memória local é comum a todos os encadeamentos do grupo, portanto, o download ocorre uma vez para o grupo e é processado por cada encadeamento no grupo e, desde a memória local é muito mais rápida que a global, os cálculos são muito mais rápidos.
Código do Kernel __kernel void ComputeBlockLocal(int offset_n1, int offset_n2, __global const nbcoord_t* mass, __global const nbcoord_t* y, __global nbcoord_t* f, int yoff, int foff, int points_count, int stride) { int n1 = get_global_id(0) + offset_n1; __global const nbcoord_t* rx = y + yoff; __global const nbcoord_t* ry = rx + stride; __global const nbcoord_t* rz = rx + 2 * stride; __global const nbcoord_t* vx = rx + 3 * stride; __global const nbcoord_t* vy = rx + 4 * stride; __global const nbcoord_t* vz = rx + 5 * stride; __global nbcoord_t* frx = f + foff; __global nbcoord_t* fry = frx + stride; __global nbcoord_t* frz = frx + 2 * stride; __global nbcoord_t* fvx = frx + 3 * stride; __global nbcoord_t* fvy = frx + 4 * stride; __global nbcoord_t* fvz = frx + 5 * stride; nbcoord_t x1 = rx[n1]; nbcoord_t y1 = ry[n1]; nbcoord_t z1 = rz[n1]; nbcoord_t res_x = 0.0; nbcoord_t res_y = 0.0; nbcoord_t res_z = 0.0; __local nbcoord_t x2[NBODY_DATA_BLOCK_SIZE]; __local nbcoord_t y2[NBODY_DATA_BLOCK_SIZE]; __local nbcoord_t z2[NBODY_DATA_BLOCK_SIZE]; __local nbcoord_t m2[NBODY_DATA_BLOCK_SIZE];
Cuda
A implementação da plataforma NVidia CUDA é um pouco mais simples que o OpenCL, não precisamos criar o contexto do dispositivo e gerenciar a fila de execução (pelo menos até que desejemos fazer uma implementação com várias GPUs). Como no caso do OpenCL, precisamos alocar memória na GPU, copiar nossos dados nela e então podemos iniciar o núcleo da computação.
Você pode ler mais sobre como trabalhar com a CUDA
aqui .
Código de inicialização do kernel CUDA dim3 grid(count / block_size); dim3 block(block_size); size_t shared_size(4 * sizeof(nbcoord_t) * block_size); kfcompute <<< grid, block, shared_size >>> (... ...);
Diferentemente do OpenCL, o CUDA não especifica toda a gama de iterações (na implementação do OpenCL é global_range), mas define o tamanho da grade e os tamanhos dos blocos na grade; portanto, o cálculo do número do corpo atual muda ligeiramente; caso contrário, o kernel é muito semelhante ao OpenCL, com exceção de outros nomes. funções de sincronização e especificador para memória compartilhada. Outro recurso distintivo útil do CUDA é que podemos especificar o tamanho necessário da memória compartilhada quando o kernel é iniciado. Como na implementação do OpenCL, no início de cada bloco de iteração, copiamos parte dos dados na memória compartilhada e, em seguida, trabalhamos com essa memória de todas as threads do bloco.
Código do kernel CUDA __global__ void kfcompute(int offset_n2, const nbcoord_t* y, int yoff, nbcoord_t* f, int foff, const nbcoord_t* mass, int points_count, int stride) { int n1 = blockDim.x * blockIdx.x + threadIdx.x; const nbcoord_t* rx = y + yoff; const nbcoord_t* ry = rx + stride; const nbcoord_t* rz = rx + 2 * stride; nbcoord_t x1 = rx[n1]; nbcoord_t y1 = ry[n1]; nbcoord_t z1 = rz[n1]; nbcoord_t res_x = 0.0; nbcoord_t res_y = 0.0; nbcoord_t res_z = 0.0; extern __shared__ nbcoord_t shared_xyzm_buf[]; nbcoord_t* x2 = shared_xyzm_buf; nbcoord_t* y2 = x2 + blockDim.x; nbcoord_t* z2 = y2 + blockDim.x; nbcoord_t* m2 = z2 + blockDim.x; for(int b2 = 0; b2 < points_count; b2 += blockDim.x) { int n2 = b2 + offset_n2 + threadIdx.x;
Tabela 3. A dependência da função de tempo de cálculo (em segundos) fn no número de corpos interagindo N para várias implementações de GPU e melhores implementações de CPU| N | 4096 | 8192 | 16384 | 32768 | 65536 | 131072 |
|---|
| openmp + block + otimização | 0,0128 | 0,0495 | 0,194 | 0,774 | --- | --- |
| OpenCL + meio NVidia K80 | 0,004 | 0,008 | 0,026 | 0,134 | 0,332 | 1,18 |
| CUDA + meia NVidia K80 | 0,004 | 0,008 | 0,0245 | 0,111 | 0,291 | 1,13 |
Onde obter NVidia K80As implementações OpenCL e CUDA foram lançadas no serviço
Colab gratuito do Google, que fornece acesso aos computadores NVidia K80. Você pode ler mais sobre como trabalhar com este serviço no
hub Em geral, o resultado é decepcionante, apenas 5-6 vezes mais rápido que a implementação da CPU. Mesmo se realizarmos cálculos em todo o K80, obteremos aceleração até 12 vezes, mas desde Como a complexidade da tarefa é quadrática, em um tempo razoável, poderemos processar não 32768 corpos em interação, mas 131072, o que é apenas 4 vezes mais.
Função Aproximação fn
Se você observar atentamente a função, definida pela força atrativa de dois corpos, fica claro que ela diminui quadraticamente com a distância. Portanto, podemos calcular com precisão a força de interação entre corpos próximos e aproximadamente entre corpos distantes. Uma abordagem famosa
é um algoritmo de
código de árvore proposto por D. Barnes e P. Hat. Uma
árvore octree é construída no espaço simulado, contendo em suas folhas as coordenadas e massas dos corpos simulados. Os nós pais contêm o centro de massa, a massa total dos nós filhos e o raio da esfera descrita ao redor dos corpos dos nós filhos. A raiz da árvore contém o centro de massa de todos os corpos, sua massa total e o raio da esfera descrita ao seu redor. Ao calcular a força de interação, a distância da raiz da árvore é considerada primeiro se a razão da distância do nó ao seu raio for maior do que alguma constante
lambdacrit , então a raiz é considerada um corpo com coordenadas iguais às coordenadas do centro de massa dos corpos incluídos nela e uma massa igual à soma das massas dos nós filhos, se a razão for menor ou igual a
lambdacrit , o procedimento é repetido recursivamente para cada nó filho. Se uma folha de uma árvore é alcançada, a força de interação é considerada da maneira usual. Assim, se um corpo está longe do grupo compacto de outros corpos, esse grupo é apresentado a ele como um corpo, e a força de interação é calculada não até cada corpo, mas apenas até um corpo. Devido a isso, a complexidade do algoritmo diminui com o
O(N2) antes
O(N log(N)) ao custo de alguma perda de precisão.
Na minha abordagem, decidi não usar a
árvore octree , mas a
árvore kd, porque é mais fácil de usar e possui menor sobrecarga de armazenamento em comparação com o octree.
Vamos voltar à implementação na CPU. Um nó da árvore kd pode ser representado na forma de uma classe que contém ponteiros para os descendentes esquerdo e direito e informações sobre as coordenadas e a massa:
Nó da árvore Kd class node { node* m_left;
Com esse método de armazenamento da árvore, temos duas opções possíveis para percorrer a árvore: use recursão explícita ou use a pilha. Eu decidi pela segunda opção.
Cálculo da força de interação atravessando uma árvore nbvertex_t force_compute(const nbvertex_t& v1, const nbcoord_t mass1) { nbvertex_t total_force; node* stack_data[MAX_STACK_SIZE] = {}; node** stack = stack_data; node** stack_head = stack; *stack++ = m_root; while(stack != stack_head) { node* curr = *--stack; const nbcoord_t distance_sqr((v1 - curr->m_mass_center).norm()); if(distance_sqr > curr->m_radius_sqr) {
Como no caso da implementação "exata" da CPU, a função de cálculo de força é chamada para cada corpo. O ciclo em todos os corpos pode ser facilmente paralelizado usando diretivas OpenMP.
Mas as iterações de loop vizinhas, neste caso, se referem a partes completamente diferentes da árvore, o que não permite o uso eficiente do cache do processador. Para superar esse problema, todos os corpos precisam ser atravessados não na ordem inicial, mas na ordem em que os corpos estão localizados nas folhas da árvore kd, iterações vizinhas ocorrerão para corpos que estão próximos no espaço e atravessarão a árvore por caminhos quase idênticos.
Travessia de folha de árvore template<class Visitor> void traverse(Visitor visit) { node* stack_data[MAX_STACK_SIZE] = {}; node** stack = stack_data; node** stack_head = stack; *stack++ = m_root; while(stack != stack_head) { node* curr = *--stack; if(curr->m_radius_sqr > 0) {
Essa implementação tem outro problema - não há uma maneira universal de paralelizar essa travessia de árvore. Mas podemos mudar completamente a maneira como a árvore é armazenada na memória - podemos armazenar todos os nós em uma matriz linear e abandonar completamente o armazenamento de ponteiros para os descendentes, por analogia com a construção de uma
pilha binária . Ao iniciar a numeração de nós com
1 se o nó estiver no número da célula
i , então o filho esquerdo dele está na cela
2i criança certa na célula
2i+1 e o pai na célula
i/2 . O nó direito correspondente à esquerda com o número
i terá um número
i+1 . A matriz em si terá um comprimento
2N e todos os nós das folhas estarão localizados no último
N Além disso, as células com espaçamento estreito serão localizadas nas células próximas da matriz. A função de passagem em árvore mudará um pouco, mas a estrutura permanece a mesma:
Cálculo de força atravessando uma árvore em uma matriz nbvertex_t force_compute(const nbvertex_t& v1, const nbcoord_t mass1) { nbvertex_t total_force; size_t stack_data[MAX_STACK_SIZE] = {}; size_t* stack = stack_data; size_t* stack_head = stack; *stack++ = NBODY_HEAP_ROOT_INDEX; while(stack != stack_head) { size_t curr = *--stack; const nbcoord_t distance_sqr((v1 - m_mass_center[curr]).norm()); if(distance_sqr > m_radius_sqr[curr]) { total_force += force(v1, m_mass_center[curr], mass1, m_mass[curr]); } else { size_t left(left_idx(curr)); size_t rght(rght_idx(curr)); if(rght < m_body_n.size()) { *stack++ = rght; } if(left < m_body_n.size()) { *stack++ = left; } } } return total_force; }
Mas essas não são todas as possibilidades que o armazenamento dos nós na matriz se abre para nós - podemos recusar a pilha ao rastrear. Para fazer isso, no ramo de código no qual vamos para os filhos do nó, adicionamos a função para calcular o próximo nó (
nextup ) e no ramo em que calculamos a força de interação, adicionamos o cálculo do próximo nó com a subárvore atual ignorada (
skipdown )
Para pular a subárvore atual, precisamos ir até a raiz (direção
D ), enquanto estamos no descendente direito, assim que alcançamos a esquerda, vamos para a subárvore direita correspondente (direção
R ), se chegarmos à raiz, a travessia da árvore será concluída.
Código da Função Ignorar Subárvore index_t skip_down(index_t idx) {
 Figura 8. Ignorando uma subárvore skipdown .
Figura 8. Ignorando uma subárvore skipdown .Para ir para o próximo nó, se possível, vá para o filho esquerdo (direção
U ) e, se não houver descendente, vá para o próximo nó 'de baixo' usando a função
skipdown .
Ir para o próximo código de função do nó index_t next_up(index_t idx, index_t tree_size) { index_t left = left_idx(idx); if(left < tree_size) { return left; } return skip_down(idx); }
 Figura 9. Transições para o próximo nó nextup .
Figura 9. Transições para o próximo nó nextup .Pode parecer que substituímos a recursão por um loop
enquanto em função
skipdown , e essa substituição não fornece nada, mas vamos ver como determinar se um nó com um número
i descendente direito. Isso pode ser feito simplesmente verificando seu número ímpar (o filho certo está na célula
2i+1 ), para isso basta calcular
i e1 . I.e. nós dividimos o número
i dois se o bit menos significativo estiver definido como um. Mas isso pode ser feito sem um loop, em muitos processadores existe uma instrução
find first set que retorna a posição do primeiro bit configurado, usando-o, transformamos o loop em quatro instruções do processador.
Código de função de subárvore de salto otimizado index_t skip_down(index_t idx) { idx = idx >> (__builtin_ffs(~idx) - 1); return left2right(idx); }
Depois disso, podemos excluir a pilha da função de deslocamento em árvore e substituí-la por um par
skipdown+próximoup , depois disso a função parece ainda mais simples:
Cálculo de força atravessando uma árvore em uma matriz sem usar uma pilha nbvertex_t force_compute(const nbvertex_t& v1, const nbcoord_t mass1) const { nbvertex_t total_force; size_t curr = NBODY_HEAP_ROOT_INDEX; size_t tree_size = m_mass_center.size(); do { const nbcoord_t distance_sqr((v1 - m_mass_center[curr]).norm()); if(distance_sqr > m_radius_sqr[curr]) { total_force += force(v1, m_mass_center[curr], mass1, m_mass[curr]); curr = skip_down(curr); } else { curr = next_up(curr, tree_size); } } while(curr != NBODY_HEAP_ROOT_INDEX); return total_force; }
No total, obtivemos seis combinações de deslocamento de árvore e cálculo de força. Compare essas abordagens em termos de tempo e qualidade de computação. Tomamos como medida de qualidade a mudança relativa na energia total do sistema após 100 iterações. Como sistema modelo, tomamos duas "galáxias" interagindo consistindo em16384corpos cada.Tabela 4. Combinações do método transversal de árvore e cálculo de força| Tipo de cálculo de deslocamento / força em árvore | Árvore com pilha | 'Pilha' com uma pilha | 'Heap' sem uma pilha |
|---|
| Iterações por número do corpo | ciclo + árvore | ciclo + pilha | cycle + heapstackless |
|---|
| Bypass de folhas | nestedtree + tree | nestedtree + heap | nestedtree + heapstackless |
|---|

| 
|
| a) Dependência do tempo de cálculo da função fn da razão entre a distância crítica e o nó da árvore e seu raio ( λcrit ) | b) A dependência da mudança relativa de energia no sistema na razão entre a distância crítica e o nó da árvore e seu raio ( λcrit ) |
| Figura 10. Resultados do cálculo da função fn de várias maneiras na CPU (número de corpos N=32768 ) |
Pode-se ver que a implementação de 'nestedtree + tree' está irremediavelmente atrasada, porque falta concorrência. Implementações com a localização de nós de árvore na matriz e a indexação como em um heap binário estão na liderança. A mudança relativa de energia é insignificante para todas as variantes comλcrit>1 .
Também na fig. 10a mostra que para (λcrit<10 ) todas as variantes do cálculo da função fn ultrapassar significativamente a velocidade da versão mais otimizada do cálculo exato ('openmp + bloco + otimização'), com um aumento adicional λcrit implementações com uma árvore perdem a versão exata.Travessal em árvore da GPU
Tentei ignorar a árvore na GPU usando a tecnologia OpenCL e CUDA. A opção de armazenar nós na forma de uma árvore foi imediatamente descartada e apenas as opções foram deixadas com o armazenamento da árvore em uma matriz com indexação como em uma pilha binária. Em geral, a implementação do núcleo de computação não é muito diferente da versão da CPU.Kernel OpenCL para aumentar a capacidade de computação atravessando uma árvore (travessia na ordem dos corpos de numeração) __kernel void ComputeTreeBH(int offset_n1, int points_count, int tree_size, __global const nbcoord_t* y, __global nbcoord_t* f, __global const nbcoord_t* tree_cmx, __global const nbcoord_t* tree_cmy, __global const nbcoord_t* tree_cmz, __global const nbcoord_t* tree_mass, __global const nbcoord_t* tree_crit_r2) { int n1 = get_global_id(0) + offset_n1; int stride = points_count; __global const nbcoord_t* rx = y; __global const nbcoord_t* ry = rx + stride; __global const nbcoord_t* rz = rx + 2 * stride; __global const nbcoord_t* vx = rx + 3 * stride; __global const nbcoord_t* vy = rx + 4 * stride; __global const nbcoord_t* vz = rx + 5 * stride; __global nbcoord_t* frx = f; __global nbcoord_t* fry = frx + stride; __global nbcoord_t* frz = frx + 2 * stride; __global nbcoord_t* fvx = frx + 3 * stride; __global nbcoord_t* fvy = frx + 4 * stride; __global nbcoord_t* fvz = frx + 5 * stride; nbcoord_t x1 = rx[n1]; nbcoord_t y1 = ry[n1]; nbcoord_t z1 = rz[n1]; nbcoord_t res_x = 0.0; nbcoord_t res_y = 0.0; nbcoord_t res_z = 0.0; int stack_data[MAX_STACK_SIZE] = {}; int stack = 0; int stack_head = stack; stack_data[stack++] = NBODY_HEAP_ROOT_INDEX; while(stack != stack_head) { int curr = stack_data[--stack]; nbcoord_t dx = x1 - tree_cmx[curr]; nbcoord_t dy = y1 - tree_cmy[curr]; nbcoord_t dz = z1 - tree_cmz[curr]; nbcoord_t r2 = (dx * dx + dy * dy + dz * dz); if(r2 > tree_crit_r2[curr]) { if(r2 < NBODY_MIN_R) { r2 = NBODY_MIN_R; } nbcoord_t r = sqrt(r2); nbcoord_t coeff = tree_mass[curr] / (r * r2); dx *= coeff; dy *= coeff; dz *= coeff; res_x -= dx; res_y -= dy; res_z -= dz; } else { int left = left_idx(curr); int rght = rght_idx(curr); if(left < tree_size) { stack_data[stack++] = left; } if(rght < tree_size) { stack_data[stack++] = rght; } } } frx[n1] = vx[n1]; fry[n1] = vy[n1]; frz[n1] = vz[n1]; fvx[n1] = res_x; fvy[n1] = res_y; fvz[n1] = res_z; }
Na primeira versão, o percurso da árvore começou na ordem de numeração dos corpos na matriz original; portanto, os threads vizinhos ignoraram partes completamente diferentes da árvore, o que afetou negativamente o desempenho do cache de memória da GPU. Portanto, na segunda modalidade, uma travessia foi aplicada, começando do topo da árvore, neste caso, os segmentos vizinhos começam a atravessar a árvore pelo mesmo caminho, porque as copas das árvores vizinhas estão próximas e no espaço. Também é importante escolhermos a numeração na matriz de nós da árvore não do zero, mas de uma, neste caso, as folhas da árvore são armazenadas na segunda metade da matriz e, com o número de corpos igual à potência de dois, teremos acesso igual à memória pelo índice tn1.Kernel do OpenCL para calcular a força atravessando uma árvore (atravessando os nós de numeração de uma árvore) __kernel void ComputeHeapBH(int offset_n1, int points_count, int tree_size, __global const nbcoord_t* y, __global nbcoord_t* f, __global const nbcoord_t* tree_cmx, __global const nbcoord_t* tree_cmy, __global const nbcoord_t* tree_cmz, __global const nbcoord_t* tree_mass, __global const nbcoord_t* tree_crit_r2, __global const int* body_n) { int tree_offset = points_count - 1 + NBODY_HEAP_ROOT_INDEX; int stride = points_count; int tn1 = get_global_id(0) + offset_n1 + tree_offset; int n1 = body_n[tn1]; nbcoord_t x1 = tree_cmx[tn1]; nbcoord_t y1 = tree_cmy[tn1]; nbcoord_t z1 = tree_cmz[tn1]; nbcoord_t res_x = 0.0; nbcoord_t res_y = 0.0; nbcoord_t res_z = 0.0; int stack_data[MAX_STACK_SIZE] = {}; int stack = 0; int stack_head = stack; stack_data[stack++] = NBODY_HEAP_ROOT_INDEX; while(stack != stack_head) { int curr = stack_data[--stack]; nbcoord_t dx = x1 - tree_cmx[curr]; nbcoord_t dy = y1 - tree_cmy[curr]; nbcoord_t dz = z1 - tree_cmz[curr]; nbcoord_t r2 = (dx * dx + dy * dy + dz * dz); if(r2 > tree_crit_r2[curr]) { if(r2 < NBODY_MIN_R) { r2 = NBODY_MIN_R; } nbcoord_t r = sqrt(r2); nbcoord_t coeff = tree_mass[curr] / (r * r2); dx *= coeff; dy *= coeff; dz *= coeff; res_x -= dx; res_y -= dy; res_z -= dz; } else { int left = left_idx(curr); int rght = rght_idx(curr); if(left < tree_size) { stack_data[stack++] = left; } if(rght < tree_size) { stack_data[stack++] = rght; } } } __global const nbcoord_t* vx = y + 3 * stride; __global const nbcoord_t* vy = y + 4 * stride; __global const nbcoord_t* vz = y + 5 * stride; __global nbcoord_t* frx = f; __global nbcoord_t* fry = frx + stride; __global nbcoord_t* frz = frx + 2 * stride; __global nbcoord_t* fvx = frx + 3 * stride; __global nbcoord_t* fvy = frx + 4 * stride; __global nbcoord_t* fvz = frx + 5 * stride; frx[n1] = vx[n1]; fry[n1] = vy[n1]; frz[n1] = vz[n1]; fvx[n1] = res_x; fvy[n1] = res_y; fvz[n1] = res_z; }
Ao percorrer a ordem de numeração dos nós da árvore, obtivemos um aumento de desempenho. Mas esta opção também pode ser melhorada. A memória global na qual os nós da árvore estão localizados atualmente é otimizada para acesso compartilhado , ou seja, threads de um grupo devem ler palavras localizadas em um bloco de memória. No nosso caso, o percurso da árvore começa nos mesmos caminhos, e solicitamos os mesmos dados com todos os threads do grupo, mas quanto mais nos aprofundamos na árvore, os caminhos dos threads vizinhos divergem cada vez mais, e precisamos solicitar dados diferentes, o que reduz o desempenho do subsistema várias vezes. memória. Mas os nós de cada subárvore pertencente ao mesmo nível estão localizados em células de memória relativamente próximas. I.e.
ao percorrer o restante da árvore, os segmentos vizinhos do núcleo de computação não acessam os mesmos nós da árvore, mas localizados na memória. Para otimizar esse acesso à memória, pode ser usada memória de textura. Mas há um problema. No momento, as texturas não suportam o trabalho com dados de precisão dupla (queremos calcular com precisão). Mas no CUDA há uma função __hiloint2double que coleta um número de precisão dupla de dois números inteiros.Código de solicitação de número de precisão dupla a partir de uma textura que armazena números inteiros template<> struct nb1Dfetch<double> { typedef double4 vec4; static __device__ double fetch(cudaTextureObject_t tex, int i) { int2 p(tex1Dfetch<int2>(tex, i)); return __hiloint2double(py, px); } static __device__ vec4 fetch4(cudaTextureObject_t tex, int i) { int ii(2 * i); int4 p1(tex1Dfetch<int4>(tex, ii)); int4 p2(tex1Dfetch<int4>(tex, ii + 1)); vec4 d4 = {__hiloint2double(p1.y, p1.x), __hiloint2double(p1.w, p1.z), __hiloint2double(p2.y, p2.x), __hiloint2double(p2.w, p2.z) }; return d4; } };
Nesse caso, duas implementações foram feitas, em uma cada elemento da árvore (x, y, z, tree_crit_r2) foi solicitado independentemente, e na segunda implementação essas solicitações foram combinadas. A solicitação para a massa do nó ocorre com muito menos frequência, apenas se a condição r2> tree_crit_r2 [curr] for atendida , portanto, não faz sentido combinar essa solicitação com as outras. Outro recurso útil da estrutura CUDA é a capacidade de controlar a proporção dos tamanhos do cache L1 e o tamanho da memória compartilhada ( cudaFuncSetCacheConfig ). No caso de passagem de árvore, não usamos memória compartilhada, portanto, podemos aumentar o cache L1 em detrimento dele.Kernel CUDA para calcular a força atravessando uma árvore (atravessando a ordem de numeração dos nós da árvore) __global__ void kfcompute_heap_bh_tex(int offset_n1, int points_count, int tree_size, nbcoord_t* f, cudaTextureObject_t tree_xyzr, cudaTextureObject_t tree_mass, const int* body_n) { nb1Dfetch<nbcoord_t> tex; int tree_offset = points_count - 1 + NBODY_HEAP_ROOT_INDEX; int stride = points_count; int tn1 = blockDim.x * blockIdx.x + threadIdx.x + offset_n1 + tree_offset; int n1 = body_n[tn1]; nbvec4_t xyzr = tex.fetch4(tree_xyzr, tn1); nbcoord_t x1 = xyzr.x; nbcoord_t y1 = xyzr.y; nbcoord_t z1 = xyzr.z; nbcoord_t res_x = 0.0; nbcoord_t res_y = 0.0; nbcoord_t res_z = 0.0; int stack_data[MAX_STACK_SIZE] = {}; int stack = 0; int stack_head = stack; stack_data[stack++] = NBODY_HEAP_ROOT_INDEX; while(stack != stack_head) { int curr = stack_data[--stack]; nbvec4_t xyzr2 = tex.fetch4(tree_xyzr, curr); nbcoord_t dx = x1 - xyzr2.x; nbcoord_t dy = y1 - xyzr2.y; nbcoord_t dz = z1 - xyzr2.z; nbcoord_t r2 = (dx * dx + dy * dy + dz * dz); if(r2 > xyzr2.w) { if(r2 < NBODY_MIN_R) { r2 = NBODY_MIN_R; } nbcoord_t r = sqrt(r2); nbcoord_t coeff = tex.fetch(tree_mass, curr) / (r * r2); dx *= coeff; dy *= coeff; dz *= coeff; res_x -= dx; res_y -= dy; res_z -= dz; } else { int left = nbody_heap_func<int>::left_idx(curr); int rght = nbody_heap_func<int>::rght_idx(curr); if(left < tree_size) { stack_data[stack++] = left; } if(rght < tree_size) { stack_data[stack++] = rght; } } } f[n1 + 3 * stride] = res_x; f[n1 + 4 * stride] = res_y; f[n1 + 5 * stride] = res_z; }
Uma análise do programa no nvprof profiler mostrou que, mesmo ao usar a memória de textura para armazenar uma árvore, ainda há uma carga muito alta na memória global.De fato, no CUDA, toda a memória do kernel endereçada a endereços 'calculados' é armazenada na memória global e, portanto, a pilha necessária para percorrer a árvore está localizada na memória global e consome uma parte significativa da largura de banda do chip de memória, porque a pilha possui cada thread de execução e há muitos threads.Mas, felizmente, já sabemos como atravessar uma árvore sem usar uma pilha. Complementando o núcleo computacional anterior com as funções de calcular o próximo nó da árvore, obtemos um novo kernel, além disso, mais compacto.Núcleo CUDA para a força da computação atravessando uma árvore sem usar uma pilha __global__ void kfcompute_heap_bh_stackless(int offset_n1, int points_count, int tree_size, nbcoord_t* f, cudaTextureObject_t tree_xyzr, cudaTextureObject_t tree_mass, const int* body_n) { nb1Dfetch<nbcoord_t> tex; int tree_offset = points_count - 1 + NBODY_HEAP_ROOT_INDEX; int stride = points_count; int tn1 = blockDim.x * blockIdx.x + threadIdx.x + offset_n1 + tree_offset; int n1 = body_n[tn1]; nbvec4_t xyzr = tex.fetch4(tree_xyzr, tn1); nbcoord_t x1 = xyzr.x; nbcoord_t y1 = xyzr.y; nbcoord_t z1 = xyzr.z; nbcoord_t res_x = 0.0; nbcoord_t res_y = 0.0; nbcoord_t res_z = 0.0; int curr = NBODY_HEAP_ROOT_INDEX; do { nbvec4_t xyzr2 = tex.fetch4(tree_xyzr, curr); nbcoord_t dx = x1 - xyzr2.x; nbcoord_t dy = y1 - xyzr2.y; nbcoord_t dz = z1 - xyzr2.z; nbcoord_t r2 = (dx * dx + dy * dy + dz * dz); if(r2 > xyzr2.w) { if(r2 < NBODY_MIN_R) { r2 = NBODY_MIN_R; } nbcoord_t r = sqrt(r2); nbcoord_t coeff = tex.fetch(tree_mass, curr) / (r * r2); dx *= coeff; dy *= coeff; dz *= coeff; res_x -= dx; res_y -= dy; res_z -= dz; curr = nbody_heap_func<int>::skip_idx(curr); } else { curr = nbody_heap_func<int>::next_up(curr, tree_size); } } while(curr != NBODY_HEAP_ROOT_INDEX); f[n1 + 3 * stride] = res_x; f[n1 + 4 * stride] = res_y; f[n1 + 5 * stride] = res_z; }
O desempenho dos núcleos em execução na GPU depende muito do tamanho dos blocos nos quais dividimos a tarefa. Depende desse tamanho, quantos registros, memória local e outros recursos estarão disponíveis para cada thread de computação. Você também deve ter em mente que, enquanto aguarda o acesso à memória em um thread, outro thread pode executar cálculos no processador de sombreador, portanto, com um número suficiente de threads em execução simultaneamente em um processador, o tempo de acesso à memória ficará oculto por trás dos cálculos. Portanto, antes de comparar o desempenho de nossos núcleos, precisamos calcular o tamanho ideal do bloco para cada um deles. Vamos fazer uma comparação na metade disponível da NVidia K80 para nós.Tabela 5. A dependência da função de tempo de cálculo (em segundos) fn GPU N=131072 λcrit=10| / | 8 | 16 | 32. | 64 | 128 | 256 | 512 | 1024 |
|---|
| opencl+dense | 5.77 | 2.84 | 1.46 | 1.13 | 1.15 | 1.14 | 1.14 | 1.13 |
| cuda+dense | 5.44 | 2.55 | 1.27 | 0.96 | 0.97 | 0.99 | 0.99 | - |
| opencl+heap+cycle | 5.88 | 5.65 | 5.74 | 5.96 | 5.37 | 5.38 | 5.35 | 5.38 |
| opencl+heap+nested | 4.54 | 3,68 | 3.98 | 5.25 | 5.46 | 5.41 | 5.48 | 5.31 |
| cuda+heap+nested | 3,62 | 2,81 | 2,68 | 4.26 | 4.84 | 4.75 | 4.8 | 4.67 |
| cuda+heap+nested+tex | 2.6 | 1.51 | 0.912 | 0.7 | 1.85 | 1.75 | 1.69 | 1.61 |
| cuda+heap+nested+tex+stackless | 2.3 | 1.29 | 0.773 | 0.5 | 0.51 | 0.52 | 0.52 | 0.52 |
6. ( ) fn GPU N=1M λcrit=4| / | 8 | 16 | 32. | 64 | 128 | 256 | 512 | 1024 |
|---|
| opencl+dense | 366 | 179 | 89.9 | 69.3 | 70.3 | 69.1 | 68.9 | 68.0 |
| cuda+dense | 346 | 162 | 79.6 | 60.8 | 60.8 | 60.7 | 59.6 | - |
| opencl+heap+cycle | 16.2 | 18.2 | 20.1 | 21.2 | 21.2 | 21.3 | 21.2 | 21.1 |
| opencl+heap+nested | 10.5 | 7.63 | 6.38 | 8.23 | 9.95 | 9.89 | 9.65 | 9.66 |
| cuda+heap+nested | 8.67 | 6.44 | 5.39 | 5.93 | 8.65 | 8.61 | 8.41 | 8.27 |
| cuda+heap+nested+tex | 6.38 | 3.57 | 2.13 | 1.44 | 3.56 | 3.46 | 3.30 | 3.29 |
| cuda+heap+nested+tex+stackless | 5.78 | 3.19 | 1.83 | 1.21 | 1.11 | 1,10 | 1.11 | 1,13 |
Uma situação difícil, mas, diferentemente da versão da CPU do percurso da árvore, é claro que cada etapa de otimização traz resultados tangíveis. A implementação do 'opencl + heap + cycle' é quase 6 vezes mais lenta que uma solução exata com cálculo de função completafn .
Uma implementação de 'opencl + heap + nested', na qual uma travessia de árvore inicia nos nós vizinhos, 1,4 vezes mais rápido que o anterior, porque A memória cache é usada melhor. Na implementação de 'cuda + heap + anested', o cache L1 foi aumentado em detrimento da memória compartilhada, o que aumentou a velocidade em 1,4 vezes, embora seja possível que na implementação do cuda o kernel de compilação seja mais otimizado (nas versões de 'opencl + denso' e 'cuda os núcleos + densos são idênticos e o desempenho da versão do cuda é ~ 1,2 vezes maior). O aumento mais significativo na velocidade computacional (3,8 vezes) é alcançado quando a árvore está localizada na memória de textura e as consultas aos elementos do nó da árvore são combinadas. A implementação com o deslocamento em árvore sem usar a pilha 'cuda + heap + nested + tex + stackless' também é 1,4 vezes mais rápida.nele, toda a largura de banda do barramento de memória é usada apenas para acessar dados sobre nós da árvore e não é gasta na pilha. Assim, comλcrit=10 Foi possível alcançar a aceleração duas vezes em comparação com o cálculo completo da função fn . Mas
λcrit=10 um valor excessivamente grande do parâmetro, no gráfico da mudança relativa de energia no sistema, da razão entre a distância crítica e o nó da árvore e seu raio para a implementação da CPU, pode ser visto que valores mais baixos podem ser usados λcritsem uma perda visível de precisão. Vamos tentar variarλcrit com os tamanhos de bloco ideais que determinamos na etapa anterior.
| 
|
a) N=128K
| b) N=1M
|
Fig. 11. Dependência do tempo de cálculo da função. fn da razão entre a distância crítica e o nó da árvore e seu raio ( λcrit ) para várias implementações de passeio em árvore da GPU
|
Pode-se ver que, para pequenas λcrit todos os métodos de cálculo de funções fnvá para valores próximos, determinados no momento da construção da árvore kd e da preparação dos dados para a GPU. Além disso, o tempo de construção de uma árvore contribui significativamente para o tempo total deλcrit≤4, esse tempo pode ser negligenciado. É interessante notar que quandoN=128K o desempenho melhora novamente ao atingir λcrit=1024provavelmente isso se deve ao fato de que todos os threads da GPU percorrem os mesmos vértices da árvore ao mesmo tempo, o que melhora o uso do cache L1 e elimina completamente a 'divergência de ramificação' . Também é visto que, com o melhor desempenho para uma implementação sem uma pilha (cuda + heap + nested + tex + stackless), ele supera a versão com a pilha em aproximadamente1.4−1.5vezes. Outras implementações são várias vezes mais lentas. Para consolidar o resultado, calcularemos o tempo em uma GPU com uma arquitetura mais nova.Resultados de lançamento da GeForce GTX 1080 Ti (precisão única)7. ( ) fn GPU N=1M λcrit=4| / | 8 | 16 | 32. | 64 | 128 | 256 | 512 | 1024 |
|---|
| opencl+dense | 47.8 | 23.4 | 11.6 | 11.59 | 12.8 | 12.8 | 12.8 | 12.8 |
| cuda+dense | 49.0 | 23.8 | 11.9 | 11.8 | 11.7 | 11.7 | 11.7 | 11.7 |
| opencl+heap+cycle | 7.48 | 8.26 | 7.73 | 7.36 | 7.33 | 7.27 | 7.25 | 7.26 |
| opencl+heap+nested | 1.33 | 1.20 | 1.41 | 2.46 | 2.53 | 2.49 | 2.44 | 2.47 |
| cuda+heap+nested | 1.23 | 1.10 | 1.31 | 2.28 | 2.29 | 2.28 | 2.23 | 2.25 |
| cuda+heap+nested+tex | 0.88 | 0.68 | 0.654 | 1.6 | 1.6 | 1.6 | 1.6 | 1.6 |
| cuda+heap+nested+tex+stackless | 0.71 | 0.47 | 0.45 | 0.43 | 0.43 | 0.42 | 0.41 | 0.395 |

|
12. fn ( λcrit ) GPU
|
Ao usar o GeForce GTX 1080 Ti para cálculo, a diferença entre as caminhadas pela árvore com uma pilha e sem uma pilha é de até duas vezes, desde que negligenciemos o tempo necessário para construir a árvore. Esse fato nos leva a garantir que, no futuro, portemos para a GPU e construamos uma árvore. Uma comparação das tabelas 5-7 mostra que não há um tamanho ideal de bloco ideal para um número diferente de corpos e, ainda mais, para diferentes arquiteturas de GPU, além disso, a diferença de tempo de cálculo pode atingir várias vezes, mesmo se você não considerar os valores de limite. Portanto, antes de cálculos demorados, faz sentido determinar o tamanho ideal do bloco para cada configuração.O principal que alcançamos é a capacidade de modelar a interação pareada de pouco mais de um milhão de corpos (220) por um tempo razoável em um, não na GPU mais recente. Em GPUs mais recentes (Tesla V100), aparentemente, será possível processar já cerca de dois milhões de corpos em interação em um tempo razoável, porque seu desempenho é cerca de quatro vezes maior que a metade do Tesla K80. Embora esse número também seja incomparável com o número de estrelas, mesmo em galáxias anãs, como a Pequena Nuvem de Magalhães , mas, na minha opinião, é impressionante.Conclusão
O uso de métodos incorporados para resolver equações diferenciais torna possível resolver problemas semelhantes com um alto nível de precisão a um custo de tempo relativamente pequeno, e a aplicação de algoritmos para aproximar a função de calcular as forças de atração aos pares nos permite processar volumes colossais de corpos em interação. Assim, diferentemente dos alienígenas das "Tarefas de três corpos", somos capazes de resolver o problemaN corpos e para um pequeno número de corpos e aglomerados de estrelas inteiros, embora ao custo de alguma perda de precisão.Visualização
Para quem leu até o fim, darei mais alguns vídeos com visualização da evolução dos sistemas dos corpos.Simulação de uma colisão de duas galáxias. O número total de corpos é de 60 mil.Modelando a evolução de uma galáxia que consiste em um milhão de estrelas. No centro está um corpo com uma massa de 99% do total. Partículas únicas são quase indistinguíveis. Já é mais como uma onda em uma gota de líquido. Colorido de acordo com o módulo de velocidade. Baixa velocidade - azul, verde - médio, vermelho - alto. Vê-se que no centro a velocidade é mais alta e diminui gradualmente para as bordas, e a mais baixa no plano equatorial.Uma simulação mais "dinâmica" da evolução de uma galáxia de milhões de estrelas. No centro está um corpo com uma massa de 10% do total. O corpo central afeta o resto significativamente menos. Primeiro, as "estrelas" se separam, depois se reúnem em vários aglomerados e, no final, formam novamente um grande aglomerado.No processo de modelagem, cerca de 5% das "estrelas" deixaram a região inicial irrevogavelmente.No décimo segundo, assemelha-se muito à galáxia de roda dentada existente .O código do projeto pode ser encontrado no github .