E se você tiver uma idéia para uma proteína fresca e saudável e quiser realizá-la? Por exemplo, você gostaria de criar uma vacina contra
H. pylori (como a
equipe eslovena da iGEM 2008 ) criando uma proteína híbrida que combina fragmentos de flagelina de
E. coli que estimulam a resposta imune com a habitual flagelina de
H. pylori ?
H. pylori Hybrid Flagellin Design Apresentado pela Equipe Eslovena no iGEM 2008Surpreendentemente, estamos muito perto de criar qualquer proteína que desejamos sem sair do notebook de Jupyter, graças aos mais recentes desenvolvimentos em genômica, biologia sintética e, mais recentemente, em laboratórios em nuvem.
Neste artigo, mostrarei o código Python da idéia de uma proteína à sua expressão em uma célula bacteriana, sem tocar uma pipeta ou conversar com alguém. O custo total será de apenas algumas centenas de dólares! Usando a
terminologia de Vijaya Pande da A16Z , isso é Biology 2.0.
Mais especificamente, no artigo, o código Python do laboratório em nuvem faz o seguinte:
- Síntese de uma sequência de DNA que codifica qualquer proteína que eu quero.
- Clonar esse DNA sintético em um vetor que possa expressá-lo.
- Transformação de bactérias com esse vetor e confirmação de que a expressão está ocorrendo.
Configuração do Python
Primeiro, as configurações gerais de Python necessárias para qualquer bloco de notas Jupyter. Importamos alguns módulos Python úteis e criamos algumas funções utilitárias, principalmente para visualização de dados.
Códigoimport re import json import logging import requests import itertools import numpy as np import seaborn as sns import pandas as pd import matplotlib as mpl import matplotlib.pyplot as plt from io import StringIO from pprint import pprint from Bio.Seq import Seq from Bio.Alphabet import generic_dna from IPython.display import display, Image, HTML, SVG def uprint(astr): print(astr + "\n" + "-"*len(astr)) def show_html(astr): return display(HTML('{}'.format(astr))) def show_svg(astr, w=1000, h=1000): SVG_HEAD = '''<?xml version="1.0" standalone="no"?><!DOCTYPE svg PUBLIC "-//W3C//DTD SVG 1.1//EN" "http://www.w3.org/Graphics/SVG/1.1/DTD/svg11.dtd">''' SVG_START = '''<svg viewBox="0 0 {w:} {h:}" version="1.1" xmlns="http://www.w3.org/2000/svg" xmlns:xlink= "http://www.w3.org/1999/xlink">''' return display(SVG(SVG_HEAD + SVG_START.format(w=w, h=h) + astr + '</svg>')) def table_print(rows, header=True): html = ["<table>"] html_row = "</td><td>".join(k for k in rows[0]) html.append("<tr style='font-weight:{}'><td>{}</td></tr>".format('bold' if header is True else 'normal', html_row)) for row in rows[1:]: html_row = "</td><td>".join(row) html.append("<tr style='font-family:monospace;'><td>{:}</td></tr>".format(html_row)) html.append("</table>") show_html(''.join(html)) def clean_seq(dna): dna = re.sub("\s","",dna) assert all(nt in "ACGTN" for nt in dna) return Seq(dna, generic_dna) def clean_aas(aas): aas = re.sub("\s","",aas) assert all(aa in "ACDEFGHIKLMNPQRSTVWY*" for aa in aas) return aas def Images(images, header=None, width="100%"):
Laboratórios em nuvem
Como a AWS ou qualquer nuvem de computação, o laboratório de nuvem possui equipamentos de biologia molecular, além de robôs que aluga pela Internet. Você pode emitir instruções para seus robôs clicando em alguns botões na interface ou escrevendo código que os programa por conta própria. Não é necessário escrever seus próprios protocolos, como farei aqui, uma parte significativa da biologia molecular são tarefas rotineiras padrão; portanto, geralmente é melhor confiar em um protocolo alienígena confiável que mostre boa interação com os robôs.
Recentemente, surgiram várias empresas com laboratórios em nuvem:
Transcriptic ,
Autodesk Wet Lab Accelerator (beta, e construído com base no Transcriptic),
Arcturus BioCloud (beta),
Emerald Cloud Lab (beta),
Synthego (ainda não foi iniciado). Existem até empresas criadas em cima de laboratórios em nuvem, como a
Desktop Genetics , especializada em CRISPR.
Artigos científicos sobre o uso de laboratórios em nuvem na ciência real estão começando a aparecer.
No momento da redação deste artigo, apenas o Transcriptic está em domínio público, portanto, o usaremos. Pelo que entendi, a maioria dos negócios da Transcriptic é criada para automatizar protocolos comuns, e escrever seus próprios protocolos em Python (como farei neste artigo) é menos comum.
 “Célula de trabalho” transcrita com refrigeradores na parte inferior e vários equipamentos de laboratório no estande
“Célula de trabalho” transcrita com refrigeradores na parte inferior e vários equipamentos de laboratório no estandeVou dar instruções aos robôs de transcrição no
protocolo automático . Autoprotocol é uma linguagem baseada em JSON para escrever protocolos para robôs de laboratório (e humanos, por assim dizer). O protocolo automático é feito principalmente
nesta biblioteca Python . O idioma foi criado originalmente e ainda é suportado pelo Transcriptic, mas, pelo que entendi, é completamente aberto. Existe uma boa
documentação .
Uma ideia interessante é que você pode escrever instruções para pessoas em laboratórios remotos na, digamos, China ou Índia, no auto-protocolo e potencialmente obter algumas vantagens ao usar pessoas (seu julgamento) e robôs (falta de julgamento). Precisamos mencionar
protocolocols.io aqui , esta é uma tentativa de padronizar protocolos para melhorar a reprodutibilidade, mas para humanos, não para robôs.
"instructions": [ { "to": [ { "well": "water/0", "volume": "500.0:microliter" } ], "op": "provision", "resource_id": "rs17gmh5wafm5p" }, ... ]
Exemplo de fragmento de protocolo automáticoConfigurações de Python para biologia molecular
Além de importar bibliotecas padrão, precisarei de alguns utilitários biológicos moleculares específicos. Este código é principalmente para protocolo automático e transcriptic.
O conceito de "volume morto" é frequentemente encontrado no código. Isso significa a última gota de líquido que os robôs da Transcriptic não conseguem tirar com uma pipeta dos tubos (porque eles não podem vê-la!). Você precisa gastar muito tempo para garantir que os frascos tenham material suficiente.
Código import autoprotocol from autoprotocol import Unit from autoprotocol.container import Container from autoprotocol.protocol import Protocol from autoprotocol.protocol import Ref
Síntese de DNA e biologia sintética
Apesar de sua conexão com a moderna biologia sintética, a síntese de DNA é uma tecnologia bastante antiga. Durante décadas, conseguimos produzir oligonucleotídeos (isto é, sequências de DNA de até 200 bases). No entanto, sempre foi caro, e a química nunca permitiu longas sequências de DNA. Recentemente, tornou-se possível, a um preço razoável, sintetizar genes inteiros (até milhares de bases). Essa conquista realmente abre a era da "biologia sintética".
A
Synthetic Genomics de Craig Venter levou a biologia sintética mais longe ao
sintetizar um organismo inteiro - com mais de um milhão de bases de comprimento. À medida que o comprimento do DNA aumenta, o problema não é mais a síntese, mas a montagem (isto é, a costura de sequências de DNA sintetizadas). Com cada montagem, você pode dobrar o comprimento do DNA (ou mais); portanto, após uma dúzia ou mais de iterações, você obtém uma
molécula bastante longa ! A distinção entre síntese e montagem deve ficar clara em breve para o usuário final.
Lei de Moore?
O preço da síntese de DNA está caindo muito rápido, de mais de US $ 0,30 há um ano, dois para cerca de US $ 0,10 hoje, mas está se desenvolvendo mais como bactérias do que como processadores. Por outro lado, os preços do seqüenciamento de DNA estão caindo mais rápido que a lei de Moore. Uma meta de
US $ 0,02 por base é definida como um ponto de inflexão onde você pode substituir muitas manipulações de DNA demoradas por síntese simples. Por exemplo, a esse preço, você pode sintetizar um plasmídeo inteiro de 3kb por
US $ 60 e pular um monte de biologia molecular. Espero que consigamos isso em alguns anos.
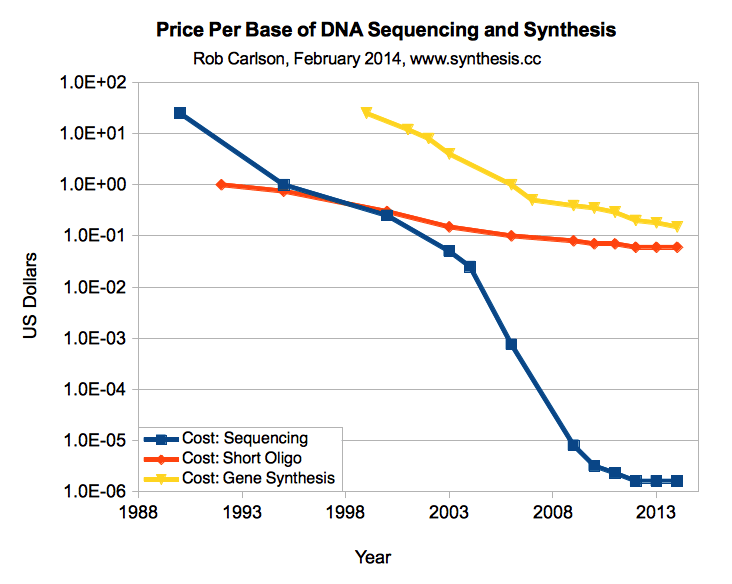 Preços de síntese de DNA em comparação com os preços de seqüenciamento de DNA, preço para 1 base (Carlson, 2014)
Preços de síntese de DNA em comparação com os preços de seqüenciamento de DNA, preço para 1 base (Carlson, 2014)Empresas de síntese de DNA
Existem várias grandes empresas no campo da síntese de DNA: O IDT é o maior produtor de oligonucleotídeos e também pode produzir "fragmentos de genes" (
gBlocks ) mais longos (até 2kb).
Gen9 ,
Twist e
DNA 2.0 geralmente se especializam em seqüências de DNA mais longas - essas são empresas de síntese de genes. Existem também algumas empresas novas e interessantes, como a
Cambrian Genomics e o
Genesis DNA , que estão trabalhando nos métodos de síntese da próxima geração.
Outras empresas, como
Amyris ,
Zymergen e
Ginkgo Bioworks , usam o DNA sintetizado por essas empresas para trabalhar no nível do corpo.
A Genomics sintética também faz isso, mas sintetiza o próprio DNA.
Recentemente, a Ginkgo
fez um acordo com a Twist para fazer 100 milhões de bases: o maior negócio que já vi. Isso prova que vivemos no futuro. A Twist chegou a anunciar um código promocional no Twitter: quando você compra 10 milhões de bases de DNA (quase todo o genoma do fermento!), Você recebe outros 10 milhões de graça.
 Oferta de nicho no Twitter Twist
Oferta de nicho no Twitter TwistParte Um: Design da Experiência
Proteína verde fluorescente
Nesta experiência, sintetizamos uma sequência de DNA para uma
proteína verde fluorescente simples (GFP). A proteína GFP foi encontrada pela primeira vez em uma
água -
viva que fluorescente sob luz ultravioleta. Esta é uma proteína extremamente útil porque é fácil detectar sua expressão simplesmente medindo a fluorescência. Existem opções de GFP que produzem amarelo, vermelho, laranja e outras cores.
É interessante ver como várias mutações afetam a cor de uma proteína, e esse é um problema de aprendizado de máquina potencialmente interessante. Mais recentemente, você precisaria gastar muito tempo no laboratório para isso, mas agora vou mostrar que é (quase) tão fácil quanto editar um arquivo de texto!
Tecnicamente, meu GFP é uma Super Folder Option (sfGFP) com algumas mutações para melhorar a qualidade.
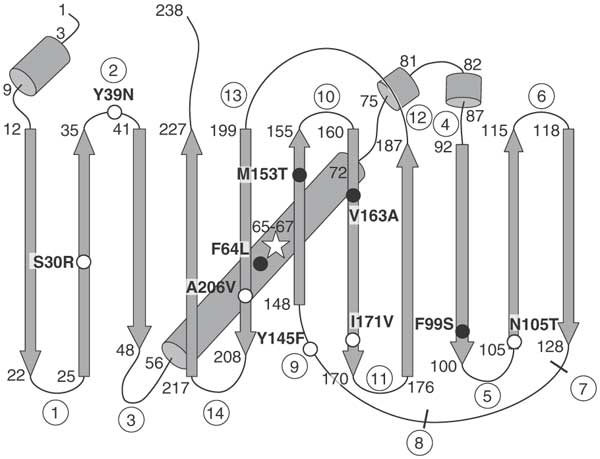 Na superfolder-GFP (sfGFP), algumas mutações fornecem determinadas propriedades úteis.
Na superfolder-GFP (sfGFP), algumas mutações fornecem determinadas propriedades úteis.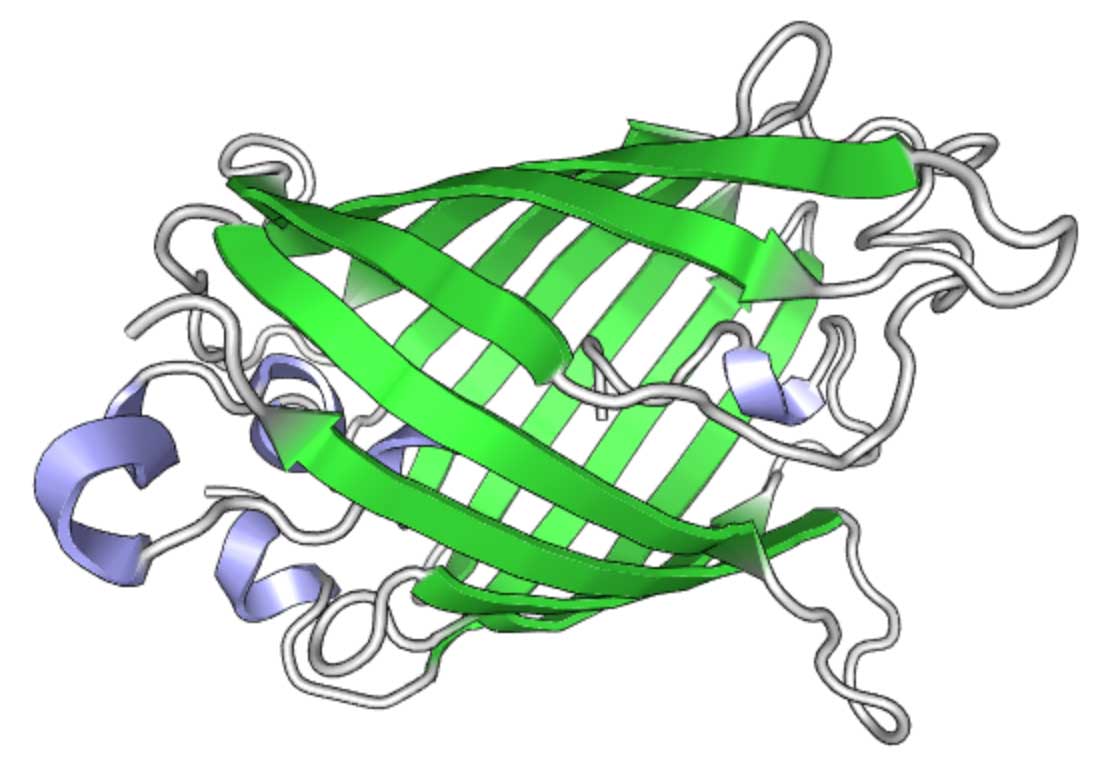 Estrutura GFP (visualizada usando PV )
Estrutura GFP (visualizada usando PV )Síntese de GFP em Twist
Eu tive a sorte de entrar no programa de testes alfa de Twist, então usei o serviço de síntese de DNA deles (eles gentilmente fizeram meu pequeno pedido - obrigado, Twist!). Esta é uma nova empresa em nosso campo, com um novo processo de síntese simplificado. Seus preços estão em torno de
US $ 0,10 por base ou menos , mas
ainda estão
na versão beta , e o programa alfa do qual participei foi fechado. A Twist levantou cerca de US $ 150 milhões, então sua tecnologia é animada.
Enviei minha sequência de DNA para o Twist como uma planilha do Excel (ainda não existe uma API, mas acho que será em breve), e eles enviaram o DNA sintetizado diretamente para minha caixa no laboratório Transcriptic (eu também usei o IDT para síntese, mas eles não enviaram DNA na Transcriptic, que estraga um pouco a diversão).
Obviamente, esse processo ainda não se tornou um caso de uso típico e requer algum suporte, mas funcionou, para que todo o pipeline permanecesse virtual. Sem isso, eu provavelmente precisaria ter acesso ao laboratório - muitas empresas não enviarão DNA ou reagentes para seu endereço residencial.
 O GFP é inofensivo, então qualquer tipo é destacado
O GFP é inofensivo, então qualquer tipo é destacadoVetor plasmídeo
Para expressar essa proteína em bactérias, o gene precisa viver em algum lugar; caso contrário, o DNA sintético que codifica o gene simplesmente se degrada instantaneamente. Como regra, em biologia molecular, usamos um plasmídeo, um pedaço de DNA redondo que vive fora do genoma bacteriano e expressa proteínas. Os plasmídeos são uma maneira conveniente de as bactérias compartilharem módulos funcionais úteis e independentes, como resistência a antibióticos. Pode haver centenas de plasmídeos em uma célula.
A terminologia amplamente usada é que um plasmídeo é um
vetor e o DNA sintético é uma inserção (inserção). Então, aqui estamos tentando clonar a inserção em um vetor e depois transformar as bactérias usando o vetor.
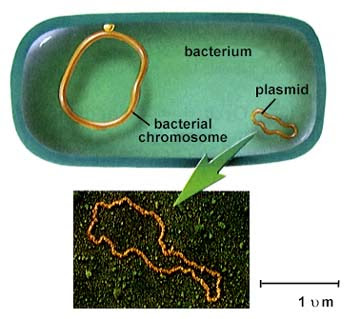 Genoma bacteriano e plasmídeo (sem escala!) ( Wikipedia )
Genoma bacteriano e plasmídeo (sem escala!) ( Wikipedia )pUC19
Eu escolhi um plasmídeo bastante padrão na série
pUC19 . Esse plasmídeo é usado com muita frequência e, como está disponível como parte do inventário Transcriptic padrão, não precisamos enviar nada a eles.
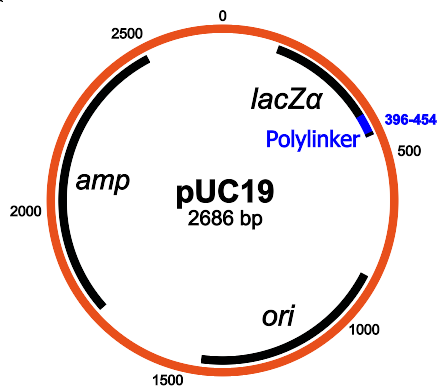 Estrutura da pUC19: os principais componentes são o gene de resistência à ampicilina, lacZα, MCS / polylinker e a origem da replicação (Wikipedia)
Estrutura da pUC19: os principais componentes são o gene de resistência à ampicilina, lacZα, MCS / polylinker e a origem da replicação (Wikipedia)A PUC19 tem uma função interessante: como contém o gene lacZα, você pode usar o método de
seleção azul e branco e ver em quais colônias a inserção foi bem-sucedida. São necessários dois produtos químicos:
IPTG e
X-gal , e o circuito funciona da seguinte maneira:
- IPTG induz a expressão de lacZα.
- Se o lacZα for desativado via DNA inserido no local de clonagem múltipla ( MCS / poliligante ) no lacZα, o plasmídeo não poderá hidrolisar X-gal e essas colônias serão brancas em vez de azuis.
- Portanto, uma inserção bem-sucedida produz colônias brancas e uma inserção com falha produz colônias azuis.
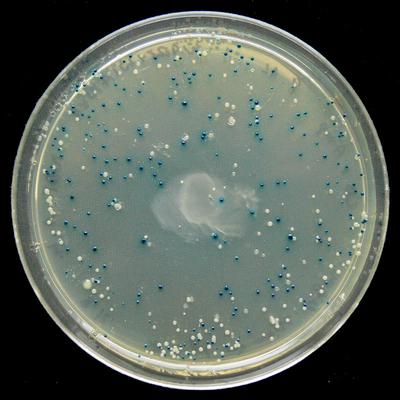 A seleção em azul e branco mostra onde a expressão lacZα foi desativada ( Wikipedia )A documentação do openwetware
A seleção em azul e branco mostra onde a expressão lacZα foi desativada ( Wikipedia )A documentação do openwetware diz:
E. coli DH5α não requer IPTG para induzir a expressão do promotor lac, mesmo se um repressor Lac for expresso na cepa. O número de cópias da maioria dos plasmídeos excede o número de repressores nas células. Se você precisar de expressão máxima, adicione IPTG a uma concentração final de 1 mM.
Sequências de DNA sintético
Sequência de DNA SfGFP
É fácil obter a sequência de DNA para sfGFP, pegando
a sequência proteica e
codificando-a com códons adequados para o organismo hospedeiro (aqui,
E. coli ). Esta é uma proteína de tamanho médio com 236 aminoácidos, portanto, a 10 centavos de síntese de DNA custa cerca de
US $ 70 por base.
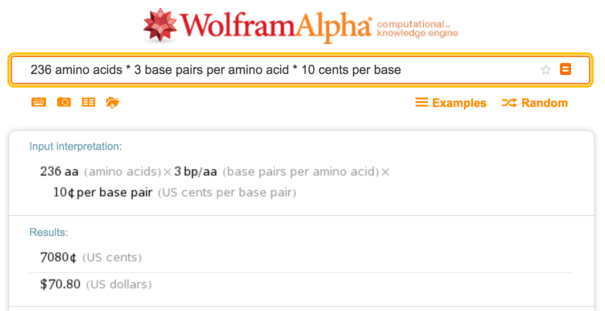 Wolfram Alpha, cálculo dos custos de síntese
Wolfram Alpha, cálculo dos custos de sínteseAs 12 primeiras bases de nosso sfGFP são a
sequência Shine-Delgarno , que eu adicionei, que teoricamente deveria aumentar a expressão (AGGAGGACAGCT, então ATG (
codão de início ) lança a proteína). De acordo com uma ferramenta computacional desenvolvida pelo
Salis Lab (
slides da aula ), podemos esperar uma expressão média a alta de nossa proteína (taxa de iniciação da tradução de 10.000 "unidades arbitrárias").
sfGFP_plus_SD = clean_seq(""" AGGAGGACAGCTATGTCGAAAGGAGAAGAACTGTTTACCGGTGTGGTTCCGATTCTGGTAGAACTGGA TGGGGACGTGAACGGCCATAAATTTAGCGTCCGTGGTGAGGGTGAAGGGGATGCCACAAATGGCAAAC TTACCCTTAAATTCATTTGCACTACCGGCAAGCTGCCGGTCCCTTGGCCGACCTTGGTCACCACACTG ACGTACGGGGTTCAGTGTTTTTCGCGTTATCCAGATCACATGAAACGCCATGACTTCTTCAAAAGCGC CATGCCCGAGGGCTATGTGCAGGAACGTACGATTAGCTTTAAAGATGACGGGACCTACAAAACCCGGG CAGAAGTGAAATTCGAGGGTGATACCCTGGTTAATCGCATTGAACTGAAGGGTATTGATTTCAAGGAA GATGGTAACATTCTCGGTCACAAATTAGAATACAACTTTAACAGTCATAACGTTTATATCACCGCCGA CAAACAGAAAAACGGTATCAAGGCGAATTTCAAAATCCGGCACAACGTGGAGGACGGGAGTGTACAAC TGGCCGACCATTACCAGCAGAACACACCGATCGGCGACGGCCCGGTGCTGCTCCCGGATAATCACTAT TTAAGCACCCAGTCAGTGCTGAGCAAAGATCCGAACGAAAAACGTGACCATATGGTGCTGCTGGAGTT CGTGACCGCCGCGGGCATTACCCATGGAATGGATGAACTGTATAAA""") print("Read in sfGFP plus Shine-Dalgarno: {} bases long".format(len(sfGFP_plus_SD))) sfGFP_aas = clean_aas("""MSKGEELFTGVVPILVELDGDVNGHKFSVRGEGEGDATNGKLTLKFICTTGKLPVPWPTLVTTLTYG VQCFSRYPDHMKRHDFFKSAMPEGYVQERTISFKDDGTYKTRAEVKFEGDTLVNRIELKGIDFKEDGNILGHKLEYNFNSHNVYITADKQKN GIKANFKIRHNVEDGSVQLADHYQQNTPIGDGPVLLPDNHYLSTQSVLSKDPNEKRDHMVLLEFVTAAGITHGMDELYK""") assert sfGFP_plus_SD[12:].translate() == sfGFP_aas print("Translation matches protein with accession 532528641")
Leia em sfGFP plus Shine-Dalgarno: 726 bases de comprimento
A tradução corresponde à proteína com a adesão 532528641
Sequência de DNA PUC19
Primeiro, verifico se a
sequência de pUC19 que baixei do NEB tem o comprimento correto e inclui o
poliligador esperado.
pUC19_fasta = !cat puc19fsa.txt pUC19_fwd = clean_seq(''.join(pUC19_fasta[1:])) pUC19_rev = pUC19_fwd.reverse_complement() assert all(nt in "ACGT" for nt in pUC19_fwd) assert len(pUC19_fwd) == 2686 pUC19_MCS = clean_seq("GAATTCGAGCTCGGTACCCGGGGATCCTCTAGAGTCGACCTGCAGGCATGCAAGCTT") print("Read in pUC19: {} bases long".format(len(pUC19_fwd))) assert pUC19_MCS in pUC19_fwd print("Found MCS/polylinker")
Leia na pUC19: 2686 bases longas
MCS / poliligante encontrado
Fazemos alguns CQs básicos para garantir que EcoRI e BamHI estejam presentes na pUC19 apenas uma vez (as seguintes enzimas de restrição estão disponíveis no inventário transcricional padrão:
PstI ,
PvuII ,
EcoRI ,
BamHI ,
BbsI ,
BsmBI ).
REs = {"EcoRI":"GAATTC", "BamHI":"GGATTC"} for rename, res in REs.items(): assert (pUC19_fwd.find(res) == pUC19_fwd.rfind(res) and pUC19_rev.find(res) == pUC19_rev.rfind(res)) assert (pUC19_fwd.find(res) == -1 or pUC19_rev.find(res) == -1 or pUC19_fwd.find(res) == len(pUC19_fwd) - pUC19_rev.find(res) - len(res)) print("Asserted restriction enzyme sites present only once: {}".format(REs.keys()))
Agora, examinamos a sequência lacZα e verificamos que não há nada inesperado. Por exemplo, deve começar com Met e terminar com um códon de parada. Também é fácil confirmar que esta é a ORF completa de 324 pb lacZα carregando a sequência de pUC19 no
visualizador de snapgene livre.
lacZ = pUC19_rev[2217:2541] print("lacZα sequence:\t{}".format(lacZ)) print("r_MCS sequence:\t{}".format(pUC19_MCS.reverse_complement())) lacZ_p = lacZ.translate() assert lacZ_p[0] == "M" and not "*" in lacZ_p[:-1] and lacZ_p[-1] == "*" assert pUC19_MCS.reverse_complement() in lacZ assert pUC19_MCS.reverse_complement() == pUC19_rev[2234:2291] print("Found MCS once in lacZ sequence")
sequência de lacZ: ATGACCATGATTACGCCAAGCTTGCATGCCTGCAGGTCGACTCTAGAGGATCCCCGGGTACCGAGCTCGAATTCACTGGCCGTCGTTTTACAACGTCGTGACTGGGAAAACCCTGGCGTTACCCAACTTAATCGCCTTGCAGCACATCCCCCTTTCGCCAGCTGGCGTAATAGCGAAGAGGCCCGCACCGATCGCCCTTCCCAACAGTTGCGCAGCCTGAATGGCGAATGGCGCCTGATGCGGTATTTTCTCCTTACGCATCTGTGCGGTATTTCACACCGCATATGGTGCACTCTCAGTACAATCTGCTCTGATGCCGCATAG
Sequência r_MCS: AAGCTTGCATGCCTGCAGGTCGACTCTAGAGGATCCCCGGGTACCGAGCTCGAATTC
MCS encontrado uma vez na sequência lacZ
Gibson assembly
Montar DNA significa simplesmente reticulação de fragmentos. Geralmente, você coleta vários fragmentos de DNA em um segmento mais longo e o clona em um plasmídeo ou genoma. Nesta experiência, quero clonar um segmento de DNA no plasmídeo pUC19 abaixo do promotor lac para expressão em
E. coli .
Existem muitos métodos de clonagem (por exemplo,
NEB ,
openwetware ,
addgene ). Aqui vou usar a montagem Gibson (
desenvolvida por Daniel Gibson na Synthetic Genomics em 2009), que não é necessariamente o método mais barato, mas simples e flexível. Você só precisa colocar o DNA que deseja coletar (com a sobreposição apropriada) em um tubo de ensaio com o Gibson Assembly Master Mix, e ele se montará!
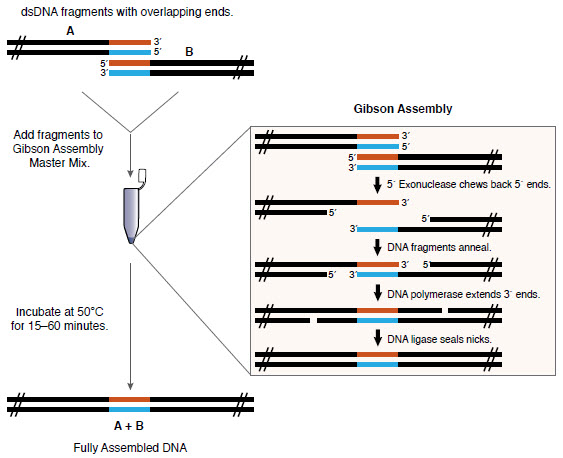 Revisão da Assembléia Gibson ( NEB )
Revisão da Assembléia Gibson ( NEB )Material de origem
Começamos com 100 ng de DNA sintético em 10 mL de líquido. Isso equivale a 0,21 picomoles de DNA ou a uma concentração de 10 ng / μl.
pmol_sfgfp = convert_ug_to_pmol(0.1, len(sfGFP_plus_SD)) print("Insert: 100ng of DNA of length {:4d} equals {:.2f} pmol".format(len(sfGFP_plus_SD), pmol_sfgfp))
Inserção: 100ng de DNA de comprimento 726 é igual a 0,21 pmol
De acordo com
o protocolo de montagem NEB , isso é material de origem suficiente:
A NEB recomenda um total de 0,02-0,5 picomoles de fragmentos de DNA quando 1 ou 2 fragmentos são montados no vetor, ou 0,2-1,0 picomoles de fragmentos de DNA quando 4-6 fragmentos são coletados.
0,02-0,5 pmoles * X μl
* A eficiência de clonagem otimizada é de 50 a 100 ng de vetores com excesso de 2-3 vezes de inserções. Use 5 vezes mais inserções se o tamanho for menor que 200 bps. O volume total de fragmentos de PCR não filtrados na reação de montagem de Gibson não deve exceder 20%.
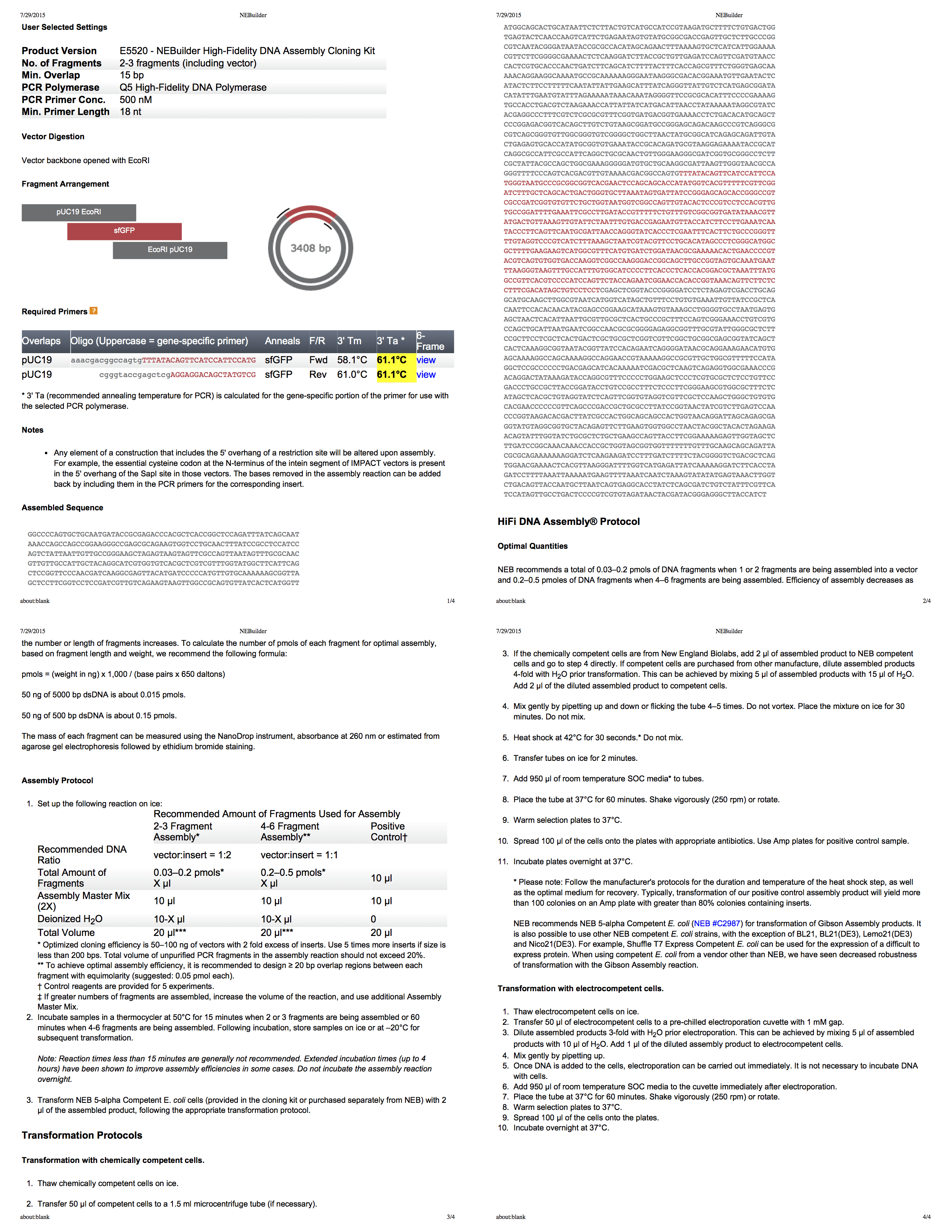
NEBuilder para montagem Gibson
O NEBuilder da Biolab é uma ferramenta realmente ótima para criar o protocolo de compilação Gibson. Ele ainda gera um PDF abrangente de quatro páginas com todas as informações. Utilizando esta ferramenta, desenvolvemos um protocolo para cortar pUC19 com EcoRI e, em seguida, usamos PCR [PCR, a reação em cadeia da polimerase permite obter um aumento significativo em pequenas concentrações de certos fragmentos de DNA no material biológico - aprox. por.] para adicionar fragmentos do tamanho apropriado à inserção.
Parte dois: experimento
O experimento consiste em quatro etapas:
- Reação de inserção em cadeia da polimerase para adicionar material com uma sequência de flanqueamento.
- Cortar um plasmídeo para acomodar a inserção.
- Montagem por inserção de Gibson e plasmídeos.
- Transformação de bactérias usando o plasmídeo montado.
Etapa 1. Inserção de PCR
A montagem de Gibson depende da sequência de DNA que você coleta, tendo alguma sequência sobreposta (consulte o protocolo NEB com instruções detalhadas acima). Além da amplificação simples, a PCR também permite adicionar uma sequência de DNA de flanqueamento, incluindo simplesmente uma sequência adicional nos primers (também pode ser clonado
usando apenas OE-PCR ).
Sintetizamos os primers de acordo com o protocolo NEB acima. Tentei
o protocolo de início rápido no site da Transcriptic, mas ainda existe
um comando de protocolo automático . A transcrição em si não sintetiza oligonucleotídeos; portanto, após 1-2 dias de espera, esses iniciadores aparecem magicamente em meu inventário (observe que a parte específica dos genes dos iniciadores é indicada em maiúscula abaixo, mas são apenas coisas cosméticas).
insert_primers = ["aaacgacggccagtgTTTATACAGTTCATCCATTCCATG", "cgggtaccgagctcgAGGAGGACAGCTATGTCG"]
Análise de Primer
Você pode analisar as propriedades desses primers usando o
IDT OligoAnalyzer .
ao depurar um experimento de PCR, é útil conhecer os pontos de fusão e a probabilidade de um efeito colateral do dímero do iniciador , embora o protocolo NEB quase certamente selecione os primers com boas propriedades. Parte do flanco específica do gene (maiúscula)
Temperatura de derretimento: 51C, 53.5C
Sequência completa
Temperatura de derretimento: 64.5C, 68.5C
Gancho de cabelo: -.4dG, -5dG
Auto-dímero: -9dG, -16dG
Heterodímero: -6dG
Eu passei por muitas iterações de PCR antes de obter resultados satisfatórios, incluindo experimentos com várias marcas diferentes de misturas de PCR. Como cada uma dessas iterações pode levar vários dias (dependendo da duração da fila do laboratório), vale a pena gastar tempo na depuração antecipada: isso economiza muito tempo a longo prazo. À medida que o poder do laboratório em nuvem aumenta, esse problema deve se tornar menos agudo. No entanto, é improvável que seu primeiro protocolo seja bem-sucedido - há muitas variáveis.Código """ PCR overlap extension of sfGFP according to NEB protocol. v5: Use 3/10ths as much primer as the v4 protocol. v6: more complex touchdown pcr procedure. The Q5 temperature was probably too hot v7: more time at low temperature to allow gene-specific part to anneal v8: correct dNTP concentration, real touchdown """ p = Protocol()
AVISO: raiz: volume baixo para o poço sfGFP 1 / sfGFP 1: 2.0: microlitro
sfGFP 1 / sfGFP 1 2.0: microlitro {'diluição': '0,25 ng / ul'}
sfgfp_pcroe_v5_puc19_primer1_10uM 75.0: microlitro {}
sfgfp_pcroe_v5_puc19_primer2_10uM 75.0: microlitro {}
Volume consolidado 52.0: microlitro
Protocol 1. Amplify the insert (oligos previously synthesized)
---------------------------------------------------------------
✓ Protocol analyzed
11 instructions
8 containers
Total Cost: $32.18
Workcell Time: $4.32
Reagents & Consumables: $27.86 : PCR
No gel, você pode avaliar o tamanho correto do produto após aumentar a concentração (a posição da tira no gel) e a quantidade correta (tira escura). O gel possui uma escada correspondente a vários comprimentos e quantidades de DNA que podem ser usados para comparação.Na fotografia de gel abaixo, as bandas D1, E1, F1 contêm, respectivamente, 2 μl, 4 μl e 8 μl do produto amplificado. Eu posso estimar a quantidade de DNA em cada faixa em comparação com o DNA na escada (50 ng de DNA por faixa na escada). Eu acho que os resultados parecem muito limpos.Tentei usar o GelEval para análise de imagens e estimativa de concentraçãoe com bastante sucesso, embora não tenha certeza se isso é muito mais preciso que o método mais ingênuo. No entanto, pequenas mudanças na localização e no tamanho das bandas levaram a grandes mudanças na estimativa da quantidade de DNA. Minha melhor estimativa da quantidade de DNA no meu produto amplificado usando GelEval é de 40 ng / μl.Supondo que estamos limitados pela quantidade de iniciador na mistura e não pela quantidade de dNTP ou enzima, uma vez que tenho 12,5 pmol de cada iniciador, isso significa um máximo teórico de 6 μg de 740bp de DNA em 25 μl. Como minha estimativa da quantidade total de DNA usando GelEval é de 40 ng x 25 μl (1 μg ou 2 pmol), esses resultados são muito razoáveis e próximos do que eu deveria esperar em condições ideais.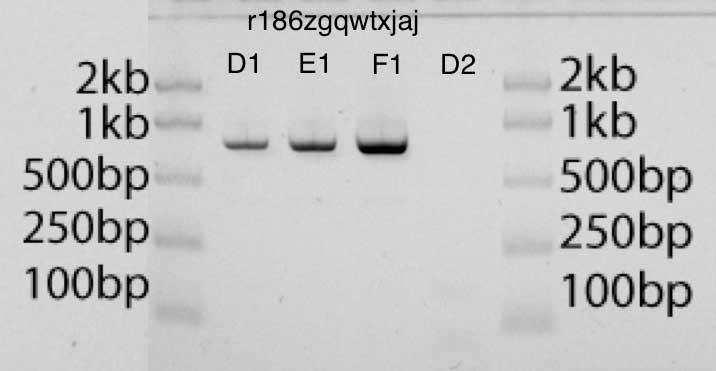 - EcoRI- pUC19, (D1, E1, F1), (D2)
- EcoRI- pUC19, (D1, E1, F1), (D2)PCR
A Transcriptic começou recentemente a fornecer dados de diagnóstico interessantes e úteis de seus robôs. No momento da redação deste artigo, eles não estavam disponíveis para download. Por enquanto, só tenho uma imagem das temperaturas durante o ciclo térmico.Os dados parecem bons, sem altos ou baixos inesperados. Um total de 35 ciclos de PCR, mas alguns desses ciclos são realizados em temperaturas muito altas como parte do touchdown da PCR . Nas minhas tentativas anteriores de amplificar esse segmento - dos quais havia vários! - houve problemas com a hibridação dos primers, então aqui a PCR trabalha por muito tempo em altas temperaturas, o que deve aumentar a precisão. Diagnósticos termocíclicos para PCR de touchdown: temperaturas de bloqueio, amostra e cobertura por 35 ciclos e 42 minutos
Diagnósticos termocíclicos para PCR de touchdown: temperaturas de bloqueio, amostra e cobertura por 35 ciclos e 42 minutosEtapa 2. Cortando o plasmídeo
Para inserir nosso DNA sfGFP em pUC19, você primeiro precisa cortar o plasmídeo. Seguindo o protocolo NEB, faço isso usando a enzima de restrição EcoRI . Existem reagentes que eu preciso no inventário padrão da Transcriptic: esse é o tampão NEB EcoRI e 10x CutSmart , bem como o plasmídeo NEB pUC19 .Para informações, abaixo estão os preços de seu inventário. De fato, pago apenas parte do preço, já que a Transcriptic aceita o pagamento pelo valor realmente consumido: ID do item Quantidade Preço de concentração
------------ ------ ------------- ----------------- - ----
CutSmart 10x B7204S 5 ml 10 X R $ 19,00
EcoRI R3101L 50.000 unidades 20.000 unidades / ml US $ 225,00
pUC19 N3041L 250 μg 1.000 μg / ml $ 268,00
Eu segui o protocolo NEB, tanto quanto possível:. 10X dH2O 1X. , , , , . 50 5 10x NEBuffer , dH2O.
, 1 λ 1 37°C 50 . , 5-10 10-20 1- .
1 50 .
Código """Protocol for cutting pUC19 with EcoRI.""" p = Protocol() experiment_name = "puc19_ecori_v3" options = {} inv = { 'water': "rs17gmh5wafm5p",
Volumes: re_tube:135.0:microliter water_tube:383.0:microliter EcoRI:30.0:microliter
Consolidated volume: 78.0:microliter
✓ Protocol analyzed
12 instructions
5 containers
Total Cost: $30.72
Tempo da célula de trabalho: $ 3,38
Reagentes e Consumíveis: $ 27,34
Resultados: corte de plasmídeo
Realizei esse experimento duas vezes sob condições ligeiramente diferentes e com géis de tamanhos diferentes, mas os resultados são quase idênticos. Eu gosto dos dois géis.Inicialmente, não aloquei espaço suficiente para o volume "morto" (em tubos de ensaio de 1,5 ml, o volume morto é de 15 µl!). Eu acho que isso explica a diferença entre D1 e E1 (as duas bandas devem ser idênticas). O problema do volume morto é fácil de resolver, criando o suprimento de trabalho adequado do EcoRI diluído no início do protocolo.Apesar desse erro, em ambos os géis, as bandas D1 e E1 têm bandas fortes na posição correta de 2,6kb. Na banda D2, um plasmídeo não cortado: como esperado, não é visível em um gel e quase invisível em outro.Duas fotos em gel parecem bem diferentes. Isso se deve em parte ao fato de que essa etapa da Transcriptic ainda não foi automatizada.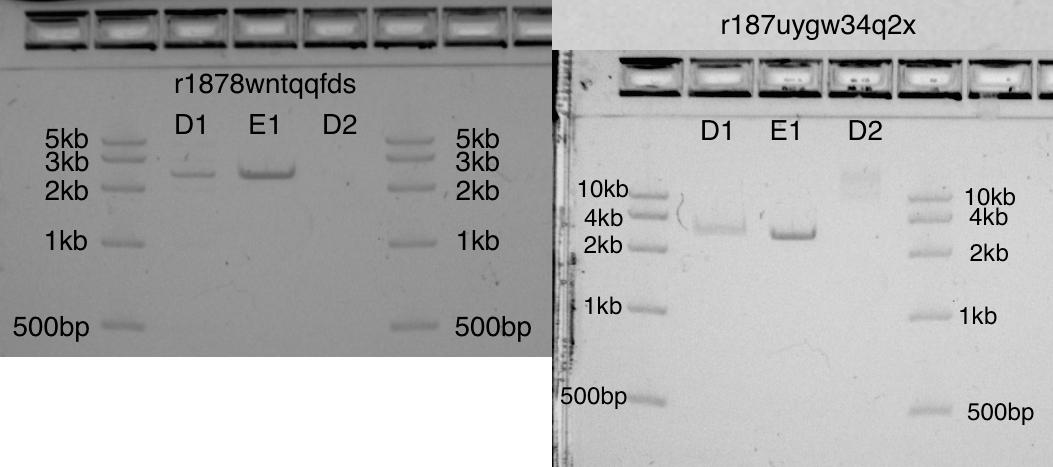 Dois géis mostrando pUC19 cortado (2,6kb) nas bandas D1 e E1 e pUC19 não cortado em D2
Dois géis mostrando pUC19 cortado (2,6kb) nas bandas D1 e E1 e pUC19 não cortado em D2Etapa 3. Montagem Gibson
A maneira mais fácil de verificar se minha montagem funciona usando o método Gibson é coletar a inserção e o plasmídeo, usar os primers M13 padrão (que flanqueiam a inserção) para amplificar parte do plasmídeo e o DNA inserido e executar qPCR e gel para garantir que a amplificação funcione. Você também pode executar uma reação de seqüenciamento para confirmar que tudo está inserido conforme o esperado, mas decidi deixá-lo para mais tarde.Se o conjunto de Gibson não funcionar, a amplificação de M13 não funcionará porque o plasmídeo foi cortado entre duas sequências M13.Código """Debugging transformation protocol: Gibson assembly followed by qPCR and a gel v2: include v3 Gibson assembly""" p = Protocol() options = {} experiment_name = "debug_sfgfp_puc19_gibson_seq_v2" inv = { "water" : "rs17gmh5wafm5p",
AVISO: raiz: volume baixo para o poço sfgfp_puc19_gibson_v1_clone / sfgfp_puc19_gibson_v1_clone: 11.0: microlitro
✓ Protocolo analisado
11 instruções
6 recipientes
Custo total: $ 32.09
Tempo de Workcell: $ 6,98
Reagentes e Consumíveis: $ 25,11
Resultados: qPCR para montagem Gibson
Eu posso acessar dados qPCR no formato JSON por meio da API Transcriptic. Esse recurso não está bem documentado , mas pode ser extremamente útil. As APIs ainda oferecem acesso a alguns dados de diagnóstico de robôs, o que pode ajudar na depuração.Primeiro, solicitamos dados de inicialização: project_id, run_id = "p16x6gna8f5e9", "r18mj3cz3fku7" api_url = "https://secure.transcriptic.com/hgbrian/{}/runs/{}/data.json".format(project_id, run_id) data_response = requests.get(api_url, headers=tsc_headers) data = data_response.json()
Em seguida, especificamos esse ID para obter os dados de "pós-processamento" do qPCR: qpcr_id = data['debug_sfgfp_puc19_gibson_seq_v1_qpcr']['id'] pp_api_url = "https://secure.transcriptic.com/data/{}.json?key=postprocessed_data".format(qpcr_id) data_response = requests.get(pp_api_url, headers=tsc_headers) pp_data = data_response.json()
Aqui estão os valores de Ct (limite do ciclo) para cada tubo. Ct é simplesmente o ponto em que a fluorescência excede um determinado valor. Ela diz aproximadamente quanto DNA existe no momento (e, portanto, aproximadamente onde começamos).
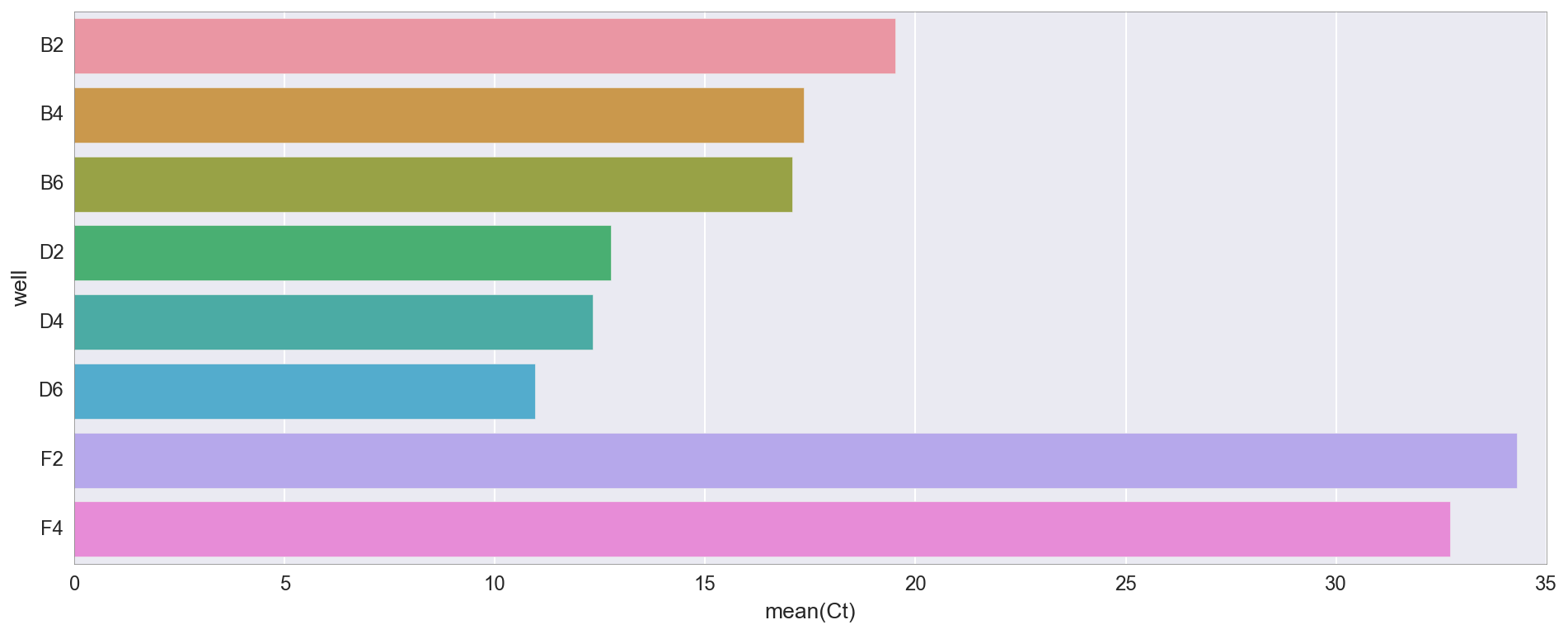 Como você pode ver, a amplificação ocorre principalmente nos tubos de teste D2 / 4/6 (onde o DNA do meu conjunto Gibson é "v3") e, em seguida, B2 / 4/6 (o conjunto Gibson é "v1"). As diferenças entre v1 e v3 são basicamente que o DNA v3 é diluído 4X de acordo com o protocolo NEB, mas ambas as opções devem funcionar. Existe alguma amplificação após o ciclo 30 em tubos de controle (F2, F4) sem modelo de DNA, mas isso não é incomum, pois inclui muito DNA iniciador.Também posso traçar a curva de amplificação de qPCR para ver a dinâmica da amplificação.
Como você pode ver, a amplificação ocorre principalmente nos tubos de teste D2 / 4/6 (onde o DNA do meu conjunto Gibson é "v3") e, em seguida, B2 / 4/6 (o conjunto Gibson é "v1"). As diferenças entre v1 e v3 são basicamente que o DNA v3 é diluído 4X de acordo com o protocolo NEB, mas ambas as opções devem funcionar. Existe alguma amplificação após o ciclo 30 em tubos de controle (F2, F4) sem modelo de DNA, mas isso não é incomum, pois inclui muito DNA iniciador.Também posso traçar a curva de amplificação de qPCR para ver a dinâmica da amplificação. f, ax = plt.subplots(figsize=(16,6)) ax.set_color_cycle(['#fb6a4a', '#de2d26', '#a50f15', '#74c476', '#31a354', '#006d2c', '#08519c', '#6baed6']) amp0 = pp_data['amp0']['SYBR']['baseline_subtracted'] _ = [plt.plot(amp0[w_n[well]], label=well) for well in ['B2', 'B4', 'B6', 'D2', 'D4', 'D6', 'F2', 'F4']] _ = ax.set_ylim(0,) _ = plt.title("qPCR (reds=Gibson v1, greens=Gibson v3, blues=control)") _ = plt.legend(bbox_to_anchor=(1, .75), bbox_transform=plt.gcf().transFigure)
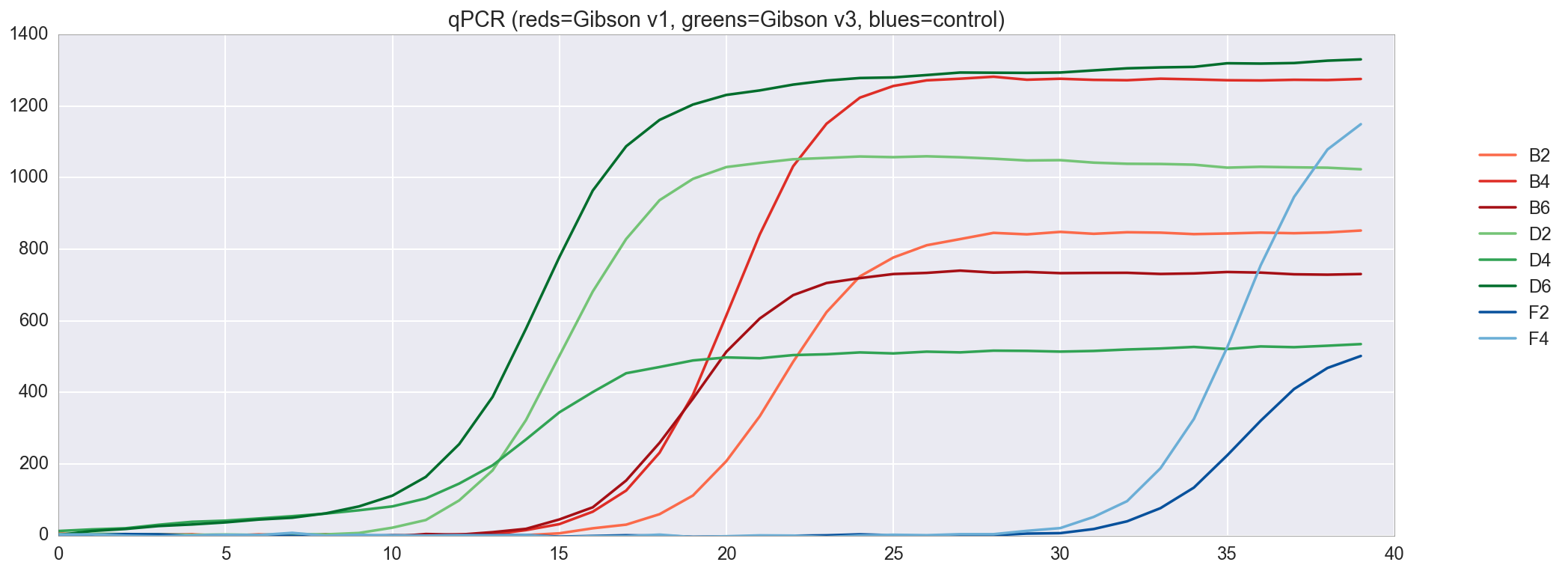 Em geral, os resultados de qPCR são ótimos, com boa amplificação de ambas as versões do meu conjunto Gibson e sem amplificação real no grupo de controle. Como o assembly v3 mostrou um resultado um pouco melhor que o v1, agora vamos usá-lo.
Em geral, os resultados de qPCR são ótimos, com boa amplificação de ambas as versões do meu conjunto Gibson e sem amplificação real no grupo de controle. Como o assembly v3 mostrou um resultado um pouco melhor que o v1, agora vamos usá-lo.Resultados: Montagem Gibson em gel
O gel também é muito limpo, mostrando bandas fortes logo abaixo de 1kb nas bandas B2, B4, B6, D2, D4, D6: esse é o tamanho que esperamos (a inserção é de cerca de 740 pb e os primers M13 têm cerca de 40 pb para cima e para baixo). A segunda faixa corresponde aos primers. Você pode ter certeza disso, pois as bandas F2 e F4 contêm apenas DNA iniciador.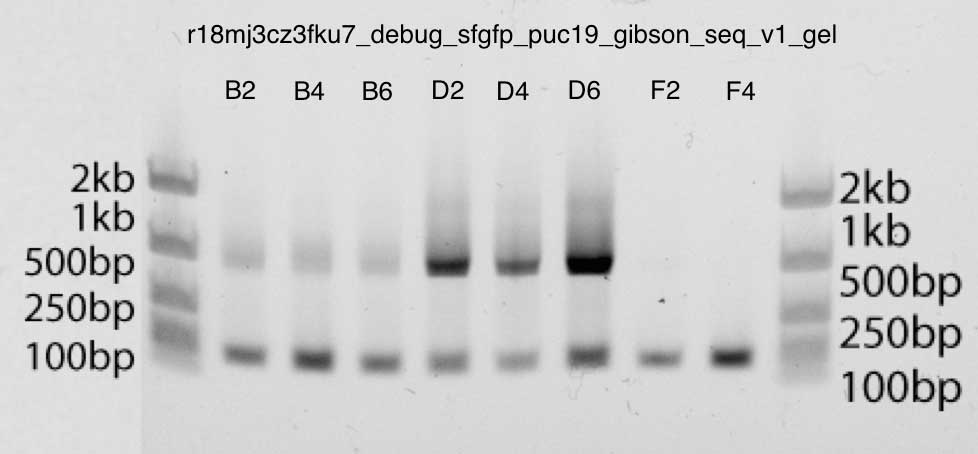 Eletroforese em gel de poliacrilamida: c O conjunto Gibson v3 mostra bandas mais fortes (D2, D4, D6), de acordo com os dados qPCR acima
Eletroforese em gel de poliacrilamida: c O conjunto Gibson v3 mostra bandas mais fortes (D2, D4, D6), de acordo com os dados qPCR acimaEtapa 4. Transformação
Transformação é o processo de mudar o corpo adicionando DNA. Nesta experiência, transformamos E. coli usando o plasmídeo pUC19 que expressa sfGFP.Utilizamos a cepa Zymo DH5α Mix & Go fácil de usar e o protocolo recomendado . Essa linhagem faz parte do inventário Transcriptic padrão. Em geral, as transformações podem ser complexas porque as células competentes são bastante frágeis; portanto, quanto mais simples e confiável o protocolo, melhor. Nos laboratórios comuns de biologia molecular, essas células competentes provavelmente seriam caras demais para uso geral. Células Zymo Mix & Go com protocolo simples
Células Zymo Mix & Go com protocolo simplesProblema com robôs
Este protocolo é um bom exemplo de como é difícil adaptar protocolos humanos para uso por robôs e como ele pode falhar inesperadamente. Os protocolos às vezes são surpreendentemente vagos ("giram o tubo de um lado para o outro"), com base no contexto geral dos biólogos moleculares, ou podem, de repente, solicitar processamento avançado de imagem ("verifique se o grânulo está misturado"). As pessoas não se importam com essas tarefas, mas os robôs precisam de instruções mais claras.Essa transformação mostrou questões interessantes de tempo. O protocolo de transformação aconselha que as células não permaneçam em temperatura ambiente por mais de alguns segundos e a placa com tubos deve ser pré-aquecida a 37 ° C. Teoricamente, você gostaria de começar o pré-aquecimento para que termine simultaneamente com a transformação, mas não está claro como os robôs Transcriptic vão lidar com essa situação - até onde eu sei, não há como sincronizar com precisão as etapas do protocolo. A falta de controle preciso do tempo parece ser um problema comum nos protocolos robóticos devido à inflexibilidade relativa do braço robótico, conflitos de agendamento, etc. Teremos que ajustar os protocolos adequadamente.Geralmente existem soluções razoáveis: às vezes você só precisa usar reagentes diferentes (por exemplo, células mais resistentes, como Mix & Go acima); às vezes você apenas hipoteca ações com uma margem (por exemplo, sacode dez vezes em vez de três); às vezes você precisa criar truques especiais para robôs (por exemplo, use uma máquina de PCR para insolação).Obviamente, a grande vantagem é que, uma vez que o protocolo funcionou uma vez, geralmente você pode confiar nele novamente. Você pode até quantificar a confiabilidade do protocolo e melhorá-lo com o tempo!Transformação de teste
Antes de iniciar a transformação com o plasmídeo totalmente montado, conduzo um experimento simples para garantir que a transformação usando o pUC19 usual (isto é, sem montagem de Gibson e sem inserção de DNA sfGFP) funcione. A pUC19 contém o gene de resistência à ampicilina; portanto, uma transformação bem-sucedida deve permitir que as bactérias cresçam em placas contendo esse antibiótico.Transfiro as bactérias diretamente para o tablet ("6 planos" na terminologia da Transcriptic), onde existe ou não ampicilina. Espero que as bactérias transformadas contenham o gene de resistência à ampicilina e, portanto, cresçam. Bactérias não transformadas não devem crescer.Código """Simple transformation protocol: transformation with unaltered pUC19""" p = Protocol() experiment_name = "debug_sfgfp_puc19_gibson_v1" inv = { "water" : "rs17gmh5wafm5p",
✓ Protocolo analisado
43 instruções
3 recipientes
$ 45,43
Resultados: transformação de teste
Nas fotografias a seguir, vemos que sem um antibiótico (placa à esquerda), observa-se crescimento nas seis placas, embora em um grau variável, o que causa preocupação. Parece que os robôs transcriptic realmente não lidam com a distribuição uniforme, o que requer certa destreza.Na presença do antibiótico (placa à direita), também há crescimento, embora novamente seja inconsistente. As duas primeiras placas com antibióticos parecem estranhas, com grande crescimento, o que é provavelmente o resultado da adição de 55 µl nessas placas, em comparação com 10 µl em placas sem antibióticos. Existem várias colônias na terceira placa e, em essência, é isso que eu esperava ver em todas as placas. Deve haver algum crescimento nas últimas três placas, mas isso não acontece. Minha única explicação para esses resultados estranhos é que eu não misturei as células e o meio o suficiente, então quase todas as células entraram nas duas primeiras placas.(Eu ainda tinha que fazer um controle positivo da ampicilina com uma bactéria inalterada, mas já fiz isso em um experimento anterior, por isso sei que a ampicilina estoque deve matar essa cepaE. coli . O crescimento é muito mais fraco nas placas de ampicilina, embora haja muito mais bactérias, como esperado).No geral, a transformação funcionou bem o suficiente para continuar, embora haja algumas falhas. Placas de células transformadas com pUC19 após 18 horas: sem antibiótico (esquerda) e com antibiótico (direita)
Placas de células transformadas com pUC19 após 18 horas: sem antibiótico (esquerda) e com antibiótico (direita)Transformação do produto após a montagem
Como a montagem de Gibson e a simples transformação de pUC19 parecem funcionar, agora você pode tentar a transformação com um plasmídeo totalmente montado que expressa sfGFP.Além da inserção coletada, também adicionarei um pouco de IPTG e X-gal às placas para ver a conversão bem-sucedida usando o método de seleção branco-azul . Esta informação adicional é útil, porque se a transformação ocorrer com a pUC19 usual, que não contém sfGFP, ela ainda dará resistência a antibióticos.Absorção e fluorescência
De acordo com esta tabela , o sfGFP brilha melhor em comprimentos de onda de excitação de 485 nm / 510 nm. Descobri que no Transcriptic, o 485/535 funciona melhor. Eu acho que porque 485 e 510 são muito semelhantes. Medo o crescimento bacteriano a 600 nm ( OD600 ).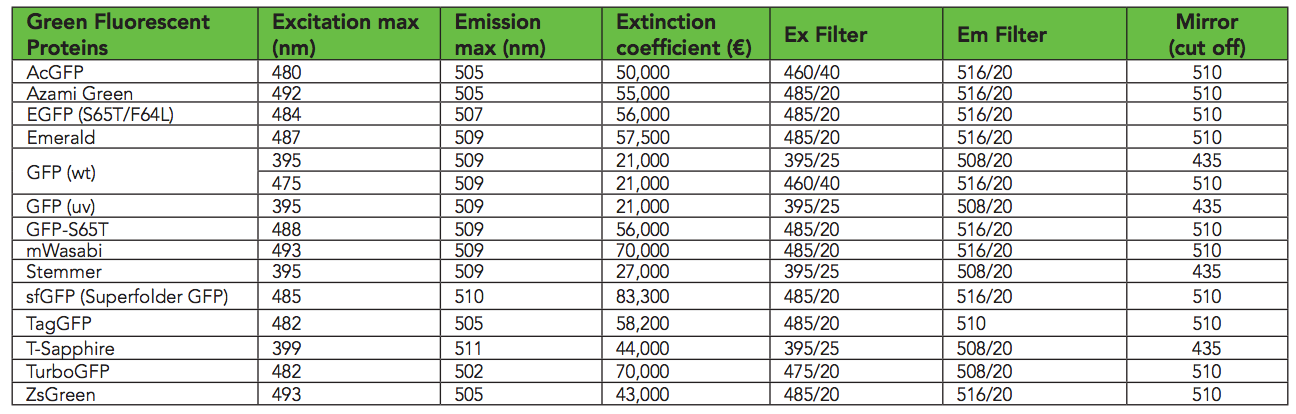 Variedade de GFP ( biotek )
Variedade de GFP ( biotek )IPTG e X-gal
Meu IPTG está na concentração de 1M e deve ser diluído 1: 1000. Por sua vez, o X-gal na concentração de 20 mg / ml também deve ser diluído 1: 1000 (20 mg / μl). Portanto, a 2000 µl de LB, adiciono 2 µl cada.De acordo com um protocolo, você deve primeiro tomar 40 mL de X-gal a uma concentração de 20 mg / ml e 40 mL de concentração de IPTG de 0,1 mM (ou 4 mL de IPTG por 1M) e incubar por 30 minutos. Esse procedimento não funcionou para mim, então apenas misturei IPTG, X-gal e as células correspondentes e usei essa mistura diretamente.Código """Full Gibson assembly and transformation protocol for sfGFP and pUC19 v1: Spread IPTG and X-gal onto plates, then spread cells v2: Mix IPTG, X-gal and cells; spread the mixture v3: exclude X-gal so I can do colony picking better v4: repeat v3 to try other excitation/emission wavelengths""" p = Protocol() options = { "gibson" : False,
Inventário: IPTG / IPTG / IPTG / IPTG / IPTG / IPTG 832.0: microlitro {}
Inventário: sfgfp_puc19_gibson_v3_clone / sfgfp_puc19_gibson_v3_clone / sfgfp_puc19_gibson_v3_clone / sfgfp_puc19_gibson_v3_clone / sfgfp_puc19_clib}
✓ Protocolo analisado
40 instruções
8 contentores
Custo total: $ 53.20
Tempo de Workcell: $ 17,35
Reagentes e Consumíveis: $ 35,86 Coleção colônia
Quando as colônias crescem em uma placa de ampicilina, eu posso "coletar" colônias individuais e plantá-las em uma placa de 96 tubos. Para isso, há uma equipe especial ( autopick ) no protocolo automático .Código """Pick colonies from plates and grow in amp media and check for fluorescence. v2: try again with a new plate (no blue colonies) v3: repeat with different emission and excitation wavelengths""" p = Protocol() options = {} for k, v in list(options.items()): if v is False: del options[k] experiment_name = "sfgfp_puc19_gibson_pick_v3" def plate_expid(val): """refer to the previous plating experiment's outputs""" plate_exp = "sfgfp_puc19_gibson_plates_v4" return "{}_{}".format(plate_exp, val)
✓ Protocolo analisado
62 instruções
8 contentores
Custo total: $ 66.38
Tempo de Trabalho: $ 57.59
Reagentes e Consumíveis: $ 8,78
Resultados: colheita de colônias
A tela azul-branca mostrou perfeitamente, principalmente, colônias brancas em placas com antibiótico (1-4) e apenas azul em placas sem antibiótico (5-6). Era exatamente o que eu esperava e fiquei feliz em vê-lo, especialmente porque usei meu próprio IPTG e X-gal, que enviei para a Transcriptic.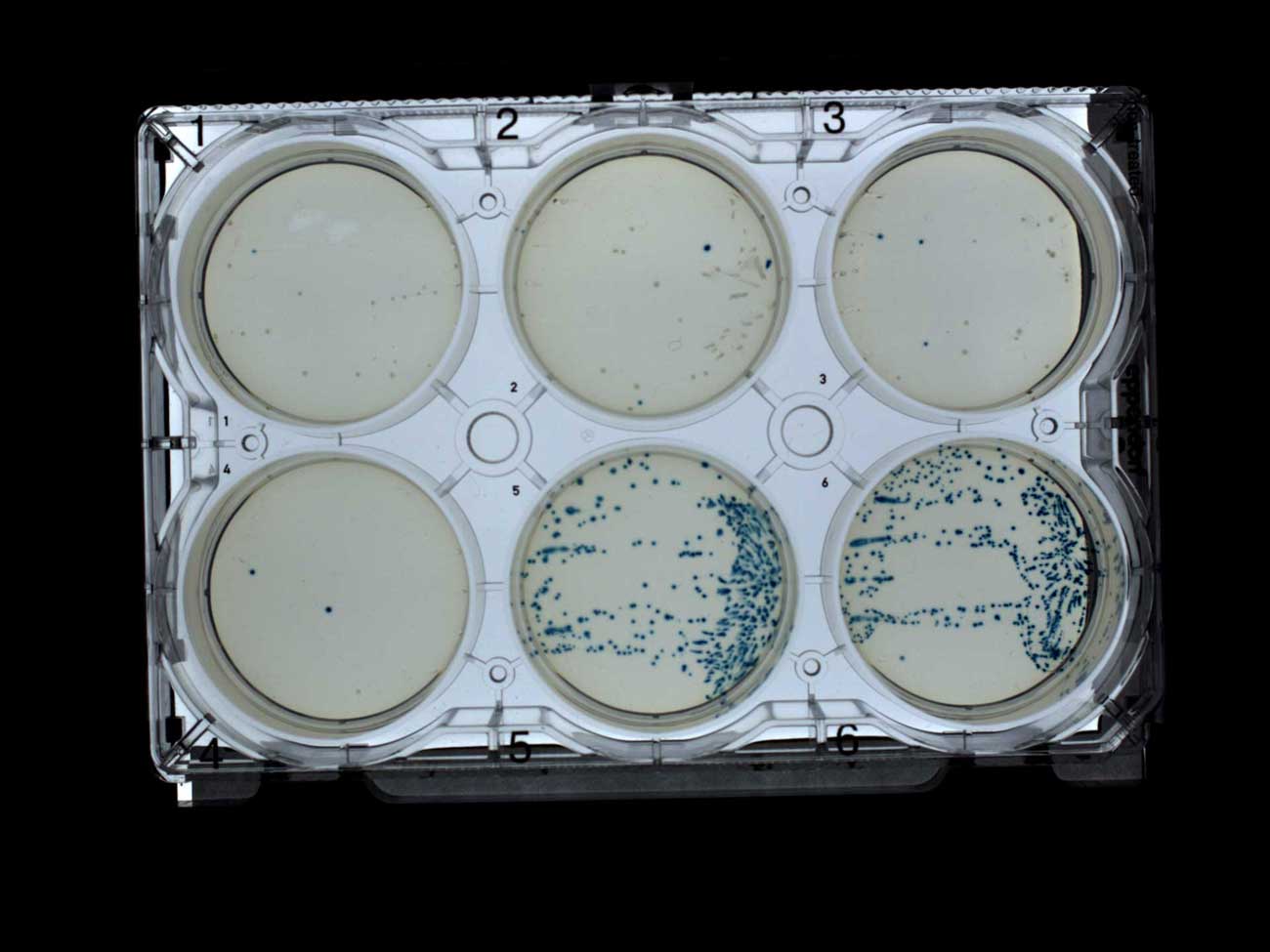 Triagem por seleção de placas azul-branco com ampicilina (1-4) e sem antibiótico (5-6)No entanto, o robô coletor de colônias não funcionou bem com essas colônias azul-brancas. A imagem abaixo foi criada subtraindo fotos sucessivas das placas após cada rodada de seleção de placas e aumentando o contraste das diferenças (no GraphicsMagick ). Dessa maneira, posso visualizar quais colônias foram coletadas (embora não idealmente, pois as colônias coletadas não são completamente removidas).Eu também assinei a imagem com o número de colônias coletadas pelo robô Transcriptic. Supunha-se que ele coletaria no máximo 10 colônias das cinco primeiras placas. No entanto, em geral, várias colônias foram coletadas, e geralmente são colônias azuis. O robô só conseguiu encontrar dez colônias em uma placa de controle com apenas colônias azuis. Minha teoria de trabalho é que um robô coletor de colônias preferencialmente coleta colônias azuis porque elas são mais contrastantes.
Triagem por seleção de placas azul-branco com ampicilina (1-4) e sem antibiótico (5-6)No entanto, o robô coletor de colônias não funcionou bem com essas colônias azul-brancas. A imagem abaixo foi criada subtraindo fotos sucessivas das placas após cada rodada de seleção de placas e aumentando o contraste das diferenças (no GraphicsMagick ). Dessa maneira, posso visualizar quais colônias foram coletadas (embora não idealmente, pois as colônias coletadas não são completamente removidas).Eu também assinei a imagem com o número de colônias coletadas pelo robô Transcriptic. Supunha-se que ele coletaria no máximo 10 colônias das cinco primeiras placas. No entanto, em geral, várias colônias foram coletadas, e geralmente são colônias azuis. O robô só conseguiu encontrar dez colônias em uma placa de controle com apenas colônias azuis. Minha teoria de trabalho é que um robô coletor de colônias preferencialmente coleta colônias azuis porque elas são mais contrastantes. Placas de triagem para seleção azul e branco com ampicilina (1-4) e sem antibiótico (5-6), indicando o número de colônias coletadasA triagem em azul e branco serviu a um propósito específico. Ele mostrou que a maioria das colônias se transforma corretamente. Pelo menos há uma inserção. No entanto, para uma melhor coleta de colônias, repeti o experimento sem o X-gal.Somente com colônias brancas o coletor de robôs conseguiu reunir dez colônias de cada uma das cinco primeiras placas. Pode-se supor que na maioria das colônias coletadas há inserções bem-sucedidas.
Placas de triagem para seleção azul e branco com ampicilina (1-4) e sem antibiótico (5-6), indicando o número de colônias coletadasA triagem em azul e branco serviu a um propósito específico. Ele mostrou que a maioria das colônias se transforma corretamente. Pelo menos há uma inserção. No entanto, para uma melhor coleta de colônias, repeti o experimento sem o X-gal.Somente com colônias brancas o coletor de robôs conseguiu reunir dez colônias de cada uma das cinco primeiras placas. Pode-se supor que na maioria das colônias coletadas há inserções bem-sucedidas. Colônias que crescem em placas com ampicilina (1-4) e sem antibiótico (5-6)
Colônias que crescem em placas com ampicilina (1-4) e sem antibiótico (5-6)Resultados: transformação com produto montado
Depois de cultivar 50 colônias selecionadas em uma placa de 96 tubos por 20 horas, medi a fluorescência para verificar a expressão de sfGFP. A Transcriptic usa um leitor Tecan Infinite para medir a fluorescência e absorção (e luminescência, se você preferir) .Em teoria, em qualquer colônia, um plasmídeo deve ser montado com crescimento, uma vez que precisa de resistência a antibióticos para crescer, e cada plasmídeo coletado expressa sfGFP. Na verdade, existem muitas razões pelas quais isso pode não ser o caso, principalmente porque você pode perder o gene sfGFP do plasmídeo sem perder a resistência à ampicilina. Uma bactéria que perde o gene sfGFP tem uma vantagem na seleção em relação a seus concorrentes, porque não gasta energia extra e, considerando um número suficiente de gerações de crescimento, isso certamente acontecerá.Coleto dados de absorção (OD600) e fluorescência a cada quatro horas por 20 horas (cerca de 60 gerações). for t in [0,4,8,12,16,20]: abs_data = pd.read_csv("glow/sfgfp_puc19_gibson_pick_v3_abs_{}.csv".format(t), index_col="Well") flr_data = pd.read_csv("glow/sfgfp_puc19_gibson_pick_v3_fl2_{}.csv".format(t), index_col="Well") if t == 0: new_data = abs_data.join(flr_data) else: new_data = new_data.join(abs_data, rsuffix='_{}'.format(t)) new_data = new_data.join(flr_data, rsuffix='_{}'.format(t)) new_data.columns = ["OD 600:nanometer_0", "Fluorescence_0"] + list(new_data.columns[2:])
Colocamos no gráfico os dados da vigésima hora e os traços das medições anteriores. De fato, estou interessado apenas nos dados mais recentes, pois é então que um pico de fluorescência deve ser observado. svg = [] W, H = 800, 500 min_x, max_x = 0, 0.8 min_y, max_y = 0, 50000 def _toxy(x, y): return W*(x-min_x)/(max_x-min_x), HH*(y-min_y)/(max_y-min_y) def _topt(x, y): return ','.join(map(str,_toxy(x,y))) ab_fls = [[row[0]] + [list(row[1])] for row in new_data.iterrows()]
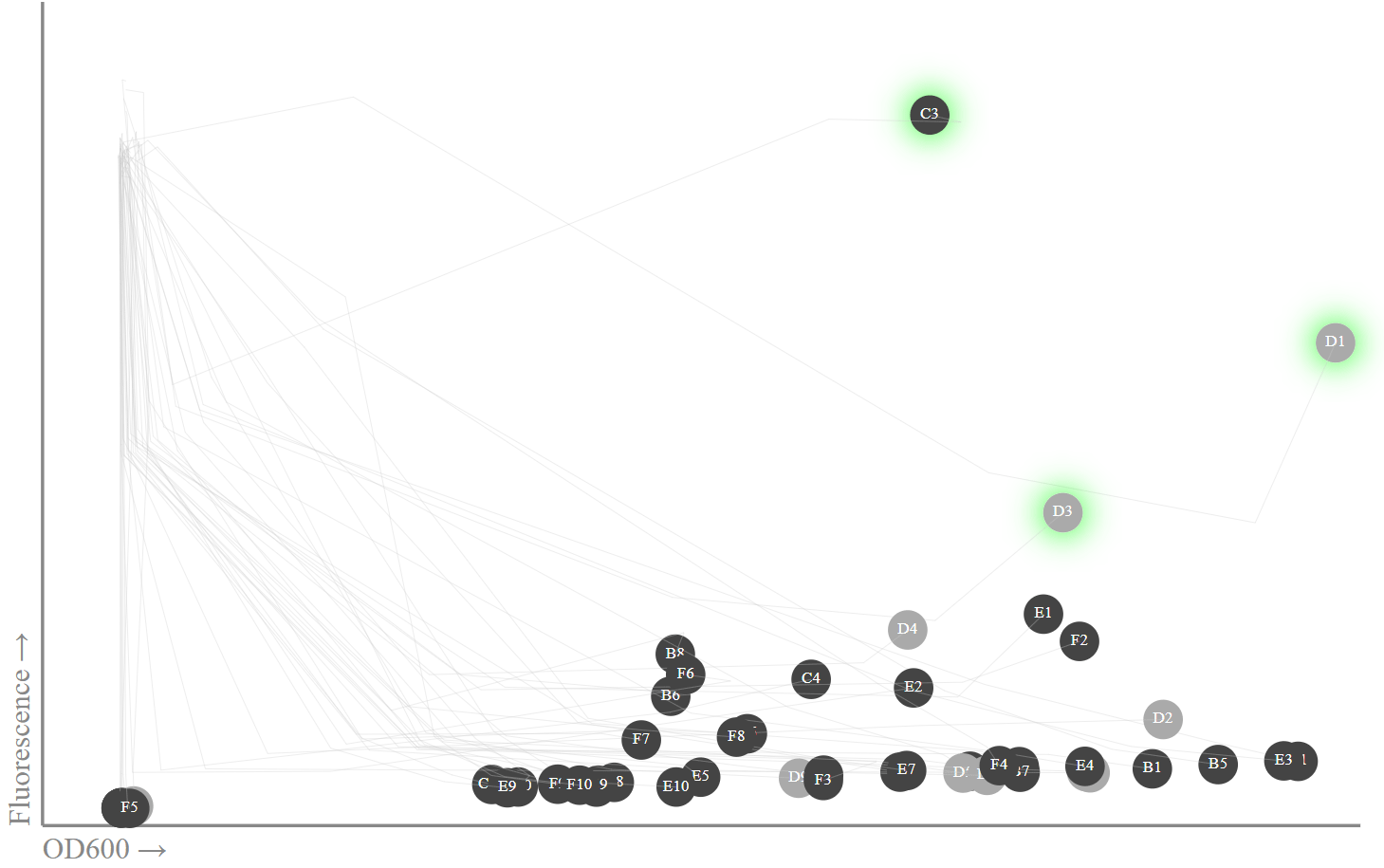 Fluorescência e OD600: colônias com ampicilina são pretas, colônias de controle sem ampicilina são cinzas. As colônias foram destacadas em verde, onde eu confirmei que a sequência da proteína sfGFP estava correta:execute o miniprep para extrair o DNA do plasmídeo e, em seguida, o sequenciamento de Sanger usando os iniciadores M13. Infelizmente, por algum motivo, o miniprep atualmente está disponível apenas pelo protocolo da web Transcriptic, e não pelo protocolo automático. Vou sequenciar os três tubos com as leituras mais altas de fluorescência (C1, D1, D3) e os outros três (B1, B3, E1), alinhando as seqüências com sfGFP usando músculo .Nos tubos C1, D3 e D3, a combinação perfeita para minha sequência sfGFP original, enquanto em B1, B3 e E1, mutações grosseiras ou alinhamento simplesmente falham.
Fluorescência e OD600: colônias com ampicilina são pretas, colônias de controle sem ampicilina são cinzas. As colônias foram destacadas em verde, onde eu confirmei que a sequência da proteína sfGFP estava correta:execute o miniprep para extrair o DNA do plasmídeo e, em seguida, o sequenciamento de Sanger usando os iniciadores M13. Infelizmente, por algum motivo, o miniprep atualmente está disponível apenas pelo protocolo da web Transcriptic, e não pelo protocolo automático. Vou sequenciar os três tubos com as leituras mais altas de fluorescência (C1, D1, D3) e os outros três (B1, B3, E1), alinhando as seqüências com sfGFP usando músculo .Nos tubos C1, D3 e D3, a combinação perfeita para minha sequência sfGFP original, enquanto em B1, B3 e E1, mutações grosseiras ou alinhamento simplesmente falham.Três colônias luminosas
Os resultados são bons, embora alguns aspectos sejam surpreendentes. Por exemplo, um leitor de fluorescência, sem motivo aparente, inicia no tempo 0 com valores muito altos (40.000 blocos). Na vigésima hora, ele se acalmou para um padrão mais razoável, com uma clara correlação entre OD600 e fluorescência (suponho que devido a uma pequena sobreposição nos espectros), além de algumas emissões com alta fluorescência. Surpreendentemente, pode ser uma, três ou, possivelmente, 11 a 15 emissões.Alguns dos tubos com altos valores de fluorescência estão localizados nos tubos de controle (ou seja, sem ampicilina, são coloridos em cinza), o que é surpreendente, uma vez que não há pressão de seleção nesses tubos, portanto, pode-se esperar perda de plasmídeo).Com base nos dados de fluorescência e nos resultados de sequenciação, parece que apenas três das 50 colônias produzem sfGFP e fluorescem. Isso não é tanto quanto eu esperava. No entanto, desde que houve três estágios de crescimento separados (in vitro, para miniprep), cerca de 200 gerações de crescimento foram submetidas a esse estágio da célula; portanto, havia muitas oportunidades de ocorrência de mutações.Deve haver maneiras de tornar o processo mais eficiente, especialmente porque estou longe de ser um especialista nesses protocolos. No entanto, produzimos com sucesso células transformadas com expressão do GFP manipulado usando apenas o código Python!Parte Três: Conclusões
Preço
Dependendo de como medir, o custo desse experimento foi de cerca de US $ 360, sem incluir o dinheiro para depuração:- US $ 70 para a síntese de DNA
- $32 PCR
- $31
- $32
- $53
- $67
- $75 3 miniprep'
Eu acho que o custo pode ser reduzido para US $ 250-300 com algumas melhorias. Por exemplo, a coleção robótica de 50 colônias é suspeita- mente cara e provavelmente pode ser abandonada.Na minha experiência, esse preço parece alto para alguns (biólogos moleculares) e baixo para outros (pessoas de TI). Como a Transcriptic basicamente cobra apenas os reagentes da lista de preços, a principal diferença nos custos é mão-de-obra. O robô já é bem barato em uma hora e ele não se importaria de acordar no meio da noite para fotografar um prato. Uma vez aprovados os protocolos, é difícil imaginar que mesmo um estudante de graduação seria mais barato, especialmente considerando os custos de oportunidade.Para maior clareza, estou apenas falando sobre a substituição de protocolos de rotina. Certamente, o desenvolvimento de protocolos avançados continuará sendo realizado por biólogos moleculares qualificados, mas muitas áreas interessantes da ciência usam protocolos rotineiros de rotina. Até recentemente, muitos laboratórios produziam seus próprios oligonucleotídeos, mas agora poucas pessoas se preocupam com isso: isso não custa a ninguém, nem mesmo estudantes de pós-graduação, quando o IDT os envia a você dentro de alguns dias.Laboratórios robóticos: prós e contras
Obviamente, eu realmente acredito no futuro dos laboratórios robóticos. Existem algumas coisas realmente engraçadas e úteis na experimentação de robôs, especialmente se você estiver envolvido principalmente na computação e for alérgico a luvas de látex e trabalho manual:- ! , . autoprotocol, .
- . 100 , .
- , , PCR. , , ? / ? , , , « 2-3 ». ?
- . . , .
- . .
- Expressividade . Você pode usar a sintaxe de programação para codificar etapas repetidas ou lógica de ramificação. Por exemplo, se você deseja dispensar 1 a 96 μl de reagente e (96 - x) μl de água em uma placa de 96 tubos, isso pode ser escrito brevemente.
- Dados legíveis por máquina . Os dados com resultados quase sempre são retornados no formato csv ou outro formato adequado ao processamento da máquina.
- Abstração . Idealmente, você pode executar o protocolo inteiro, independentemente dos reagentes utilizados ou do estilo de clonagem e substituir algo, se necessário, se funcionar melhor.
Obviamente, existem algumas desvantagens, especialmente desde que as ferramentas começaram a se desenvolver. Se compararmos com a Internet, estaremos na área de 1994:- Transcriptic — . , , , . , , .
- — Transcriptic.
- , . Transcriptic ( , , ).
- Para muitos laboratórios, pode ser mais caro usar um laboratório em nuvem do que apenas levar um estudante de pós-graduação para fazer o trabalho (custo marginal por hora: ~ $ 0). Depende se o laboratório precisa das mãos do aluno ou de sua inteligência.
- A transcrição ainda não está fazendo experiências no fim de semana. Você pode entendê-los, mas isso pode ser inconveniente, mesmo se você tiver um projeto pequeno.
Software para fabricação de proteínas
Embora exista muito código e muita depuração, acho que é possível criar algum tipo de software que use uma sequência de proteínas como entrada e crie bactérias com a expressão dessa proteína na saída.Para que isso funcione, várias coisas devem acontecer:- Verdadeira integração do Twist / IDT / Gen9 ao Transcriptic (provavelmente será lento devido à baixa demanda atual).
- , , , , . .
- ( NEB, IDT) (, primer3 ).
Em muitas aplicações, você também deseja purificar sua proteína (através de uma coluna ), ou talvez apenas fazer com que as bactérias a secretem . Suponha que em breve possamos fazer isso em um laboratório em nuvem, ou que possamos realizar experimentos in vivo (ou seja, dentro de uma célula bacteriana).Existem muitas possibilidades para o protocolo realmente funcionar melhor que os seres humanos, por exemplo: o design de promotores e o RBS para otimizar a expressão específica da sua sequência; estatísticas da probabilidade de sucesso de um experimento com base em experimentos comparáveis; análise automatizada de gel.Por que tudo isso?
Depois de tudo isso, pode não estar completamente claro por que criar essa proteína. Aqui estão algumas idéias:- - //, .
- , , .
- in vivo split-GFP .
- scFv . scFvs - .
- BiTE , ( , ).
- Faça uma vacina local que entre no corpo através dos folículos capilares (eu não recomendo tentar isso em casa).
- Mutagenize sua proteína de centenas de maneiras diferentes e veja o que acontece. Então escalar para 1000 ou 10.000 mutações? Talvez caracterizar mutações GFP?
Para novas idéias sobre o que é possível com o design de proteínas, veja centenas de projetos do iGEM .No final, quero agradecer à Transcriptic Ben Miles por sua ajuda na conclusão deste projeto.