Como ler este artigo : Peço desculpas pelo fato de o texto ter sido tão longo e caótico. Para economizar seu tempo, começo cada capítulo com a introdução de "O que aprendi", no qual explico a essência do capítulo em uma ou duas frases.
"Apenas mostre a solução!" Se você quiser apenas ver o que eu cheguei, vá para o capítulo "Torne-se mais inventivo", mas acho mais interessante e útil ler sobre falhas.Recentemente, fui instruído a configurar um processo para processar um grande volume das seqüências de DNA originais (tecnicamente, esse é um chip SNP). Era necessário obter rapidamente dados sobre um determinado local genético (chamado SNP) para modelagem subsequente e outras tarefas. Com a ajuda do R e do AWK, consegui limpar e organizar os dados de maneira natural, acelerando bastante o processamento de solicitações. Isso não foi fácil para mim e exigiu inúmeras iterações. Este artigo ajudará você a evitar alguns dos meus erros e a demonstrar o que fiz no final.
Primeiro, algumas explicações introdutórias.
Dados
Nosso Centro de Processamento de Informações Genéticas da Universidade nos forneceu 25 TB de dados TSV. Dividi-os em 5 pacotes compactados pelo Gzip, cada um contendo cerca de 240 arquivos de quatro gigabytes. Cada linha continha dados para um SNP de uma pessoa. No total, foram transmitidos dados sobre ~ 2,5 milhões de SNPs e ~ 60 mil pessoas. Além das informações do SNP, havia numerosas colunas nos arquivos com números refletindo várias características, como intensidade de leitura, frequência de diferentes alelos etc. Havia cerca de 30 colunas com valores únicos.
Finalidade
Como em qualquer projeto de gerenciamento de dados, o mais importante era determinar como os dados seriam usados. Nesse caso,
na maioria das vezes, selecionaremos modelos e fluxos de trabalho para o SNP com base no SNP . Ou seja, ao mesmo tempo, precisaremos de dados para apenas um SNP. Eu tive que aprender a extrair todos os registros relacionados a um dos 2,5 milhões de SNPs da maneira mais simples possível, mais rápida e barata.
Como não fazer
Vou citar um clichê adequado:
Não falhei mil vezes, acabei de descobrir mil maneiras de não analisar um monte de dados em um formato conveniente para consultas.
Primeira tentativa
O que aprendi : não há uma maneira barata de analisar 25 TB por vez.
Depois de ouvir o assunto "Métodos avançados de processamento de Big Data" na Universidade de Vanderbilt, tive certeza de que era um chapéu. Talvez demore uma ou duas horas para configurar o servidor Hive para executar todos os dados e informar o resultado. Como nossos dados são armazenados no AWS S3, usei o serviço
Athena , que permite aplicar consultas do Hive SQL aos dados do S3. Não é necessário configurar / aumentar o cluster do Hive e pagar apenas pelos dados que você está procurando.
Depois de mostrar a Athena meus dados e seu formato, executei alguns testes com consultas semelhantes:
select * from intensityData limit 10;
E rapidamente obteve resultados bem estruturados. Feito.
Até tentarmos usar os dados no trabalho ...
Pediram-me para extrair todas as informações do SNP para testar o modelo. Eu executei uma consulta:
select * from intensityData where snp = 'rs123456';
... e esperou. Após oito minutos e mais de 4 TB dos dados solicitados, obtive o resultado. Athena cobra uma taxa pela quantidade de dados encontrada, a US $ 5 por terabyte. Portanto, essa solicitação única custa US $ 20 e oito minutos de espera. Para executar o modelo de acordo com todos os dados, foi necessário esperar 38 anos e pagar US $ 50 milhões, o que obviamente não nos convinha.
Era necessário usar Parquet ...
O que aprendi : Tenha cuidado com o tamanho dos seus arquivos Parquet e com a organização deles.
No começo, tentei corrigir a situação convertendo todos os TSVs em
arquivos Parquet . Eles são convenientes para trabalhar com grandes conjuntos de dados, porque as informações neles são armazenadas em forma de coluna: cada coluna está em seu próprio segmento de memória / disco, diferente dos arquivos de texto nos quais as linhas contêm elementos de cada coluna. E se você precisar encontrar algo, basta ler a coluna necessária. Além disso, um intervalo de valores é armazenado em cada arquivo de uma coluna; portanto, se o valor desejado não estiver no intervalo da coluna, o Spark não perderá tempo digitalizando o arquivo inteiro.
Executei uma tarefa simples do
AWS Glue para converter nossos TSVs em Parquet e soltei novos arquivos no Athena. Demorou cerca de 5 horas. Mas quando lancei a solicitação, demorou quase o mesmo tempo e um pouco menos de dinheiro para concluí-la. O fato é que o Spark, tentando otimizar a tarefa, simplesmente desempacotou um pedaço de TSV e o colocou em seu próprio pedaço de parquet. E como cada pedaço era grande o suficiente e continha os registros completos de muitas pessoas, todos os SNPs eram armazenados em cada arquivo; portanto, o Spark precisava abrir todos os arquivos para extrair as informações necessárias.
Curiosamente, o tipo de compactação padrão (e recomendado) no Parquet - snappy - não é divisível. Portanto, cada executor manteve a tarefa de descompactar e baixar o conjunto de dados completo de 3,5 GB.

Entendemos o problema
O que aprendi : a classificação é difícil, principalmente se os dados forem distribuídos.
Pareceu-me que agora eu entendia a essência do problema. Tudo o que eu precisava fazer era classificar os dados por coluna SNP, não por pessoas. Em seguida, vários SNPs serão armazenados em um bloco de dados separado, e a função inteligente do Parquet "abrir apenas se o valor estiver no intervalo" se manifestará em toda a sua glória. Infelizmente, classificar bilhões de linhas espalhadas por um cluster provou ser uma tarefa assustadora.
A AWS certamente não deseja devolver o dinheiro por causa de "eu sou um estudante distraído". Depois que comecei a classificar no Amazon Glue, ele funcionou por 2 dias e caiu.
E o particionamento?
O que eu aprendi : Partições no Spark devem ser equilibradas.
Então surgiu a idéia de particionar os dados nos cromossomos. Existem 23 deles (e mais alguns, devido ao DNA mitocondrial e áreas não mapeadas).
Isso permitirá que você divida os dados em porções menores. Se você adicionar apenas uma linha
partition_by = "chr" à função de exportação Spark no script Glue, os dados deverão ser classificados em buckets.
 O genoma consiste em vários fragmentos chamados cromossomos.
O genoma consiste em vários fragmentos chamados cromossomos.Infelizmente, isso não funcionou. Os cromossomos têm tamanhos diferentes e, portanto, uma quantidade diferente de informações. Isso significa que as tarefas que o Spark enviou aos trabalhadores não foram balanceadas e executadas lentamente, porque alguns nós foram concluídos anteriormente e estavam inativos. No entanto, as tarefas foram concluídas. Mas, ao solicitar um SNP, o desequilíbrio novamente causou problemas. O custo do processamento de SNPs em cromossomos maiores (ou seja, de onde queremos obter os dados) diminuiu apenas 10 vezes. Muito, mas não o suficiente.
E se você se dividir em partições ainda menores?
O que eu aprendi : nunca tente fazer 2,5 milhões de partições.
Decidi dar um passeio e particionei todos os SNP. Isso garantiu o mesmo tamanho de partições.
RUIM ERA UMA IDEIA . Aproveitei o Glue e adicionei a linha
partition_by = 'snp' inocente. A tarefa iniciou e começou a executar. Um dia depois, verifiquei e vi que nada estava escrito no S3 até agora; então, matei a tarefa. Parece que o Glue estava gravando arquivos intermediários em um local oculto no S3, e muitos arquivos, talvez alguns milhões. Como resultado, meu erro custou mais de mil dólares e não agradou meu mentor.
Particionamento + classificação
O que aprendi : a classificação ainda é difícil, assim como a instalação do Spark.
A última tentativa de particionamento foi que particionei os cromossomos e depois classifiquei cada partição. Em teoria, isso aceleraria cada solicitação, porque os dados SNP desejados deveriam estar dentro de vários blocos do Parquet dentro de um determinado intervalo. Infelizmente, classificar até os dados particionados provou ser uma tarefa difícil. Como resultado, mudei para o EMR para um cluster personalizado e usei oito instâncias poderosas (C5.4xl) e Sparklyr para criar um fluxo de trabalho mais flexível ...
# Sparklyr snippet to partition by chr and sort w/in partition # Join the raw data with the snp bins raw_data group_by(chr) %>% arrange(Position) %>% Spark_write_Parquet( path = DUMP_LOC, mode = 'overwrite', partition_by = c('chr') )
... no entanto, a tarefa ainda não foi concluída. Eu sintonizei de todas as formas: aumentei a alocação de memória para cada executor de consulta, usei nós com uma grande quantidade de memória, usei variáveis de transmissão, mas cada vez que isso se transformou em meias medidas e gradualmente os executores começaram a falhar, até que tudo parasse.
Estou ficando mais inventivo
O que aprendi : algumas vezes dados especiais requerem soluções especiais.
Cada SNP tem um valor de posição. Este é o número correspondente ao número de bases ao longo de seu cromossomo. Essa é uma maneira boa e natural de organizar nossos dados. No começo, eu queria dividir por região de cada cromossomo. Por exemplo, posições 1 - 2000, 2001 - 4000, etc. Mas o problema é que os SNPs não são distribuídos igualmente entre os cromossomos, e é por isso que o tamanho dos grupos varia muito.
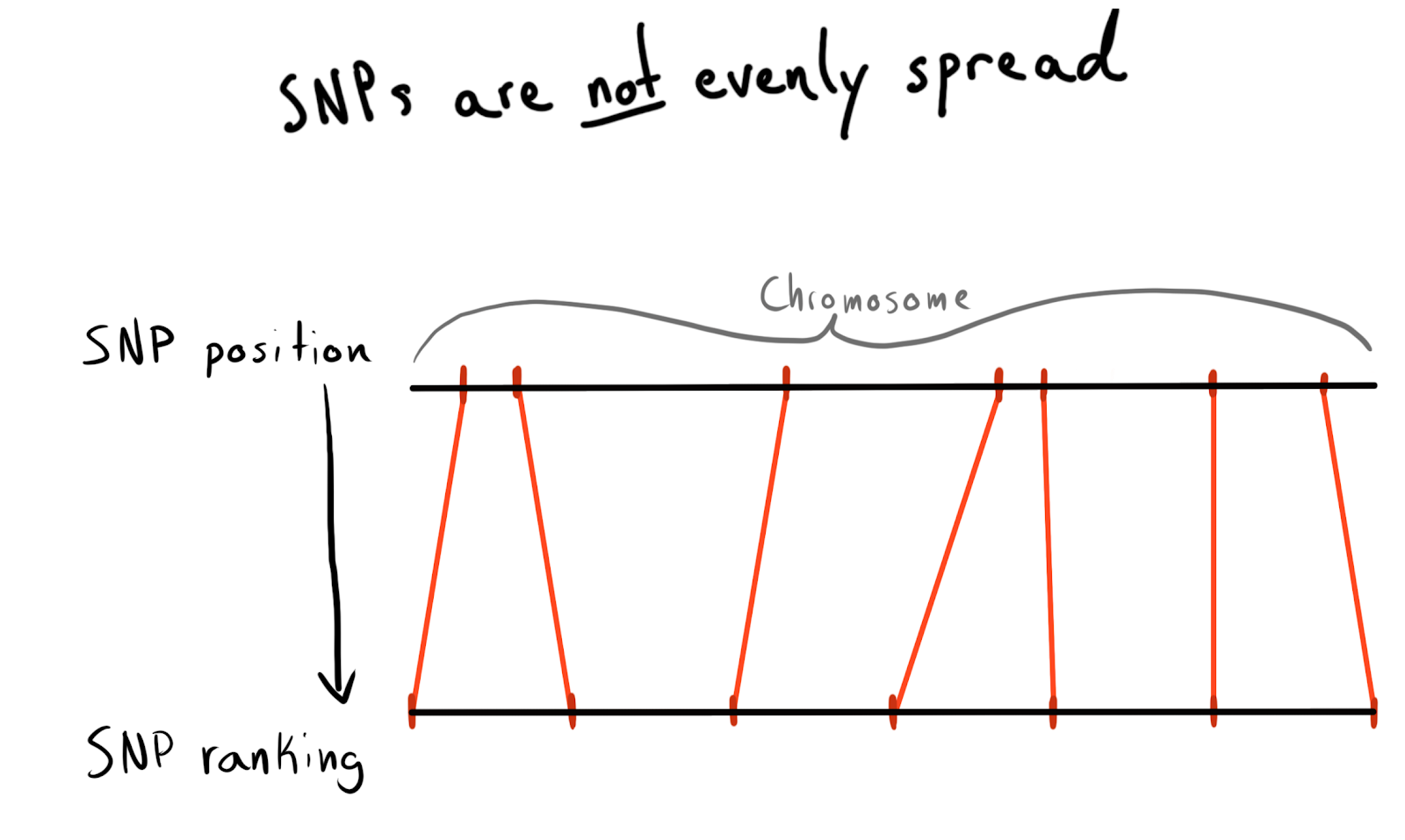
Como resultado, vim para ser dividido em categorias (posições). De acordo com os dados já baixados, solicitei uma lista de SNPs exclusivos, suas posições e cromossomos. Depois, ele classificou os dados dentro de cada cromossomo e coletou o SNP em grupos (bin) de um determinado tamanho. Diga 1000 SNP cada. Isso me deu um relacionamento SNP com um grupo no cromossomo.
No final, criei grupos (bin) no SNP 75, explicarei o motivo abaixo.
snp_to_bin <- unique_snps %>% group_by(chr) %>% arrange(position) %>% mutate( rank = 1:n() bin = floor(rank/snps_per_bin) ) %>% ungroup()
Primeira tentativa com Spark
O que aprendi : a integração do Spark é rápida, mas o particionamento ainda é caro.
Eu queria ler esse pequeno quadro de dados (2,5 milhões de linhas) no Spark, combiná-lo com dados brutos e depois particionar pela coluna
bin recém-adicionada.
Como usei
sdf_broadcast() , o Spark descobre que deve enviar um quadro de dados para todos os nós. Isso é útil se os dados forem pequenos e necessários para todas as tarefas. Caso contrário, o Spark tenta ser inteligente e distribui os dados conforme necessário, o que pode causar freios.
E, novamente, minha ideia não funcionou: as tarefas funcionaram por um tempo, concluíram a fusão e, como os executores lançados pelo particionamento, começaram a falhar.
Adicionar AWK
O que aprendi : não durma quando o básico lhe ensinar. Certamente alguém já resolveu seu problema nos anos 80.
Até o momento, a causa de todas as minhas falhas no Spark foi a confusão de dados no cluster. Talvez a situação possa ser melhorada pelo pré-processamento. Decidi tentar dividir os dados de texto bruto em colunas de cromossomos, então esperava fornecer ao Spark dados "pré-particionados".
Pesquisei no StackOverflow como descobrir os valores das colunas e encontrei
uma resposta ótima. Usando o AWK, você pode dividir um arquivo de texto em valores de coluna gravando no script, em vez de enviar os resultados para o
stdout .
Para testar, escrevi um script Bash. Baixei um dos TSVs compactados, descompactei-o com o
gzip e enviei-o para o
awk .
gzip -dc path/to/chunk/file.gz | awk -F '\t' \ '{print $1",..."$30">"chunked/"$chr"_chr"$15".csv"}'
Funcionou!
Enchimento do núcleo
O que eu aprendi :
gnu parallel é uma coisa mágica, todos deveriam usá-lo.
A separação foi bastante lenta e, quando iniciei o
htop para testar o uso de uma instância poderosa (e cara) do EC2, verificou-se que eu estava usando apenas um núcleo e aproximadamente 200 MB de memória. Para resolver o problema e não perder muito dinheiro, foi necessário descobrir como paralelizar o trabalho. Felizmente, na impressionante
Data Science de Jeron Janssens
no livro
Command Line , encontrei um capítulo sobre paralelização. Com isso, aprendi sobre o
gnu parallel , um método muito flexível para implementar multithreading no Unix.
Quando iniciei a partição usando um novo processo, tudo estava bem, mas havia um gargalo - o download de objetos S3 para o disco não era muito rápido e não era completamente paralelo. Para consertar isso, eu fiz o seguinte:
- Descobri que é possível implementar a etapa de download do S3 diretamente no pipeline, eliminando completamente o armazenamento intermediário no disco. Isso significa que posso evitar a gravação de dados brutos no disco e usar um armazenamento ainda menor e, portanto, mais barato na AWS.
- O
aws configure set default.s3.max_concurrent_requests 50 comandos aws configure set default.s3.max_concurrent_requests 50 aumentou muito o número de threads que a CLI da AWS usa (há 10 por padrão).
- Mudei para a instância do EC2 otimizada para a velocidade da rede, com a letra n no nome. Descobri que a perda de poder de computação ao usar n-instâncias é mais do que compensada por um aumento na velocidade de download. Para a maioria das tarefas, usei o c5n.4xl.
pigz gzip para pigz , esta é uma ferramenta gzip que pode fazer coisas legais para paralelizar a tarefa inigualável de descompactar arquivos (isso ajudou o mínimo).
Essas etapas são combinadas entre si para que tudo funcione muito rapidamente. Graças à maior velocidade de download e à rejeição da gravação em disco, agora eu podia processar um pacote de 5 terabytes em apenas algumas horas.
Este tweet deveria mencionar 'TSV'. Infelizmente.
Usando dados analisados novamente
O que aprendi : O Spark adora dados não compactados e não gosta de combinar partições.
Agora, os dados estavam no S3 em um formato descompactado (lido, compartilhado) e semi-ordenado, e eu poderia retornar ao Spark novamente. Uma surpresa me esperou: novamente não consegui o desejado! Era muito difícil dizer ao Spark exatamente como os dados foram particionados. E mesmo quando eu fiz isso, verificou-se que havia muitas partições (95 mil), e quando reduzi seu número a limites coerentes com
coalesce , isso arruinou minha partição. Estou certo de que isso pode ser corrigido, mas em alguns dias de pesquisa não consegui encontrar uma solução. No final, concluí todas as tarefas no Spark, apesar de levar algum tempo, e meus arquivos Parquet divididos não eram muito pequenos (~ 200 Kb). No entanto, os dados eram onde eram necessários.
 Muito pequeno e diferente, maravilhoso!
Muito pequeno e diferente, maravilhoso!Testando solicitações locais do Spark
O que eu aprendi : o Spark tem muita sobrecarga na solução de problemas simples.
Ao baixar os dados em um formato inteligente, pude testar a velocidade. Configurei um script no R para iniciar o servidor Spark local e carreguei o quadro de dados Spark no repositório especificado dos grupos Parquet (bin). Tentei carregar todos os dados, mas não consegui que o Sparklyr reconhecesse o particionamento.
sc <- Spark_connect(master = "local") desired_snp <- 'rs34771739' # Start a timer start_time <- Sys.time() # Load the desired bin into Spark intensity_data <- sc %>% Spark_read_Parquet( name = 'intensity_data', path = get_snp_location(desired_snp), memory = FALSE ) # Subset bin to snp and then collect to local test_subset <- intensity_data %>% filter(SNP_Name == desired_snp) %>% collect() print(Sys.time() - start_time)
A execução levou 29,415 segundos. Muito melhor, mas não muito bom para testar qualquer coisa em massa. Além disso, não consegui acelerar o trabalho com o cache, porque quando eu tentava armazenar em cache o quadro de dados na memória, o Spark sempre travava, mesmo quando eu alocava mais de 50 GB de memória para um conjunto de dados com peso inferior a 15.
Voltar ao AWK
O que eu aprendi : matrizes associativas AWK são muito eficientes.
Eu entendi que poderia alcançar uma velocidade maior. Lembrei que no excelente guia AWK de
Bruce Barnett , li sobre um recurso interessante chamado "
matrizes associativas ". Na verdade, esses são pares de valores-chave, que por algum motivo foram chamados de maneira diferente no AWK, e, portanto, de alguma forma não os mencionei particularmente.
Roman Cheplyaka lembrou que o termo "matrizes associativas" é muito mais antigo que o termo "par de valores-chave". Mesmo se você
pesquisar o valor-chave no Google Ngram , você não verá esse termo lá, mas encontrará matrizes associativas! Além disso, o par de valores-chave é mais frequentemente associado aos bancos de dados, portanto, é muito mais lógico comparar com o hashmap. Percebi que poderia usar essas matrizes associativas para conectar meus SNPs à tabela bin e aos dados brutos sem usar o Spark.
Para isso, no script AWK, usei o bloco
BEGIN . Este é um pedaço de código que é executado antes da primeira linha de dados ser transferida para o corpo principal do script.
join_data.awk BEGIN { FS=","; batch_num=substr(chunk,7,1); chunk_id=substr(chunk,15,2); while(getline < "snp_to_bin.csv") {bin[$1] = $2} } { print $0 > "chunked/chr_"chr"_bin_"bin[$1]"_"batch_num"_"chunk_id".csv" }
O comando
while(getline...) carregou todas as linhas do grupo CSV (bin), defina a primeira coluna (nome do SNP) como a chave para a matriz associativa
bin e o segundo valor (group) como valor. Em seguida, no bloco
{ } , aplicado a todas as linhas do arquivo principal, cada linha é enviada ao arquivo de saída, que recebe um nome exclusivo, dependendo do grupo (bin):
..._bin_"bin[$1]"_...As
chunk_id e
chunk_id correspondiam aos dados fornecidos pelo pipeline, o que evitava o estado de corrida, e cada encadeamento de execução iniciado por
parallel gravava em seu próprio arquivo exclusivo.
Como eu espalhei todos os dados brutos em pastas nos cromossomos deixados após minha experiência anterior com o AWK, agora eu poderia escrever outro script Bash para processar no cromossomo de uma vez e fornecer dados particionados mais profundos para o S3.
DESIRED_CHR='13'
O script tem duas seções
parallel .
A primeira seção lê dados de todos os arquivos que contêm informações sobre o cromossomo desejado e, em seguida, esses dados são distribuídos por fluxos que espalham arquivos nos grupos correspondentes (bin). Para impedir que condições de corrida ocorram quando vários fluxos são gravados no mesmo arquivo, o AWK transfere os nomes dos arquivos para gravação de dados em locais diferentes, por exemplo,
chr_10_bin_52_batch_2_aa.csv . Como resultado, muitos arquivos pequenos são criados no disco (para isso, usei volumes EBS de terabyte).
O pipeline da segunda seção
parallel percorre os grupos (bin) e combina seus arquivos individuais em CSVs comuns com
cat , e os envia para exportação.
Transmitir para R?
O que eu aprendi : você pode acessar
stdin e
stdout partir de um script R e, portanto, usá-lo no pipeline.
No script Bash, você pode observar esta linha:
...cat chunked/*_bin_{}_*.csv | ./upload_as_rds.R... ...cat chunked/*_bin_{}_*.csv | ./upload_as_rds.R... Ele traduz todos os arquivos de grupo concatenados (bin) no script R abaixo.
{} é uma técnica
parallel especial que insere qualquer dado enviado por ele no fluxo especificado diretamente no próprio comando. A opção
{#} fornece um ID de encadeamento exclusivo e
{%} representa o número do slot de trabalho (repetido, mas nunca ao mesmo tempo). Uma lista de todas as opções pode ser encontrada na
documentação. #!/usr/bin/env Rscript library(readr) library(aws.s3) # Read first command line argument data_destination <- commandArgs(trailingOnly = TRUE)[1] data_cols <- list(SNP_Name = 'c', ...) s3saveRDS( read_csv( file("stdin"), col_names = names(data_cols), col_types = data_cols ), object = data_destination )
Quando a variável
file("stdin") é passada para
readr::read_csv , os dados traduzidos no script R são carregados no quadro, que são gravados diretamente no S3 como um arquivo
aws.s3 usando o
aws.s3 .
O RDS é um pouco como uma versão mais nova do Parquet, sem os detalhes do armazenamento em coluna.
Depois de concluir o script Bash, recebi
.rds arquivos
.rds no S3, o que me permitiu usar tipos eficientes de compactação e
.rds .
Apesar de usar o freio R, tudo funcionou muito rápido. Não é de surpreender que os fragmentos em R responsáveis pela leitura e gravação de dados sejam bem otimizados. Após o teste em um cromossomo de tamanho médio, a tarefa foi concluída na instância C5n.4xl em cerca de duas horas.
Limitações do S3
O que aprendi : graças à implementação inteligente de caminhos, o S3 pode processar muitos arquivos.
Eu estava preocupado se o S3 poderia lidar com muitos arquivos transferidos para ele. Eu poderia tornar os nomes dos arquivos significativos, mas como o S3 os procurará?
 S3 ,
S3 , / . FAQ- S3., S3 - . (bucket) , — .
Amazon, , «-----» . : get-, . , 20 . bin-. , , (, , ). .
?
: — .
: « ?» ( gzip CSV- 7 ) . , R Parquet ( Arrow) Spark. R, , , .
: , .
, .
EC2 , ( , Spark ). , , AWS- 10 .
R .
S3 , .
library(aws.s3) library(tidyverse) chr_sizes <- get_bucket_df( bucket = '...', prefix = '...', max = Inf ) %>% mutate(Size = as.numeric(Size)) %>% filter(Size != 0) %>% mutate(
, , ,
num_jobs , .
num_jobs <- 7
purrr .
1:1000 %>% map_df(shuffle_job) %>% filter(sd == min(sd)) %>% pull(data) %>% pluck(1)
, . Bash-
for . 10 . , . , .
for DESIRED_CHR in "16" "9" "7" "21" "MT" do
:
sudo shutdown -h now
… ! AWS CLI
user_data Bash- . , .
aws ec2 run-instances ...\ --tag-specifications "ResourceType=instance,Tags=[{Key=Name,Value=<<job_name>>}]" \ --user-data file://<<job_script_loc>>
!
: API .
- . , . API .
.rds Parquet-, , . R-.
, ,
get_snp .
pkgdown , .
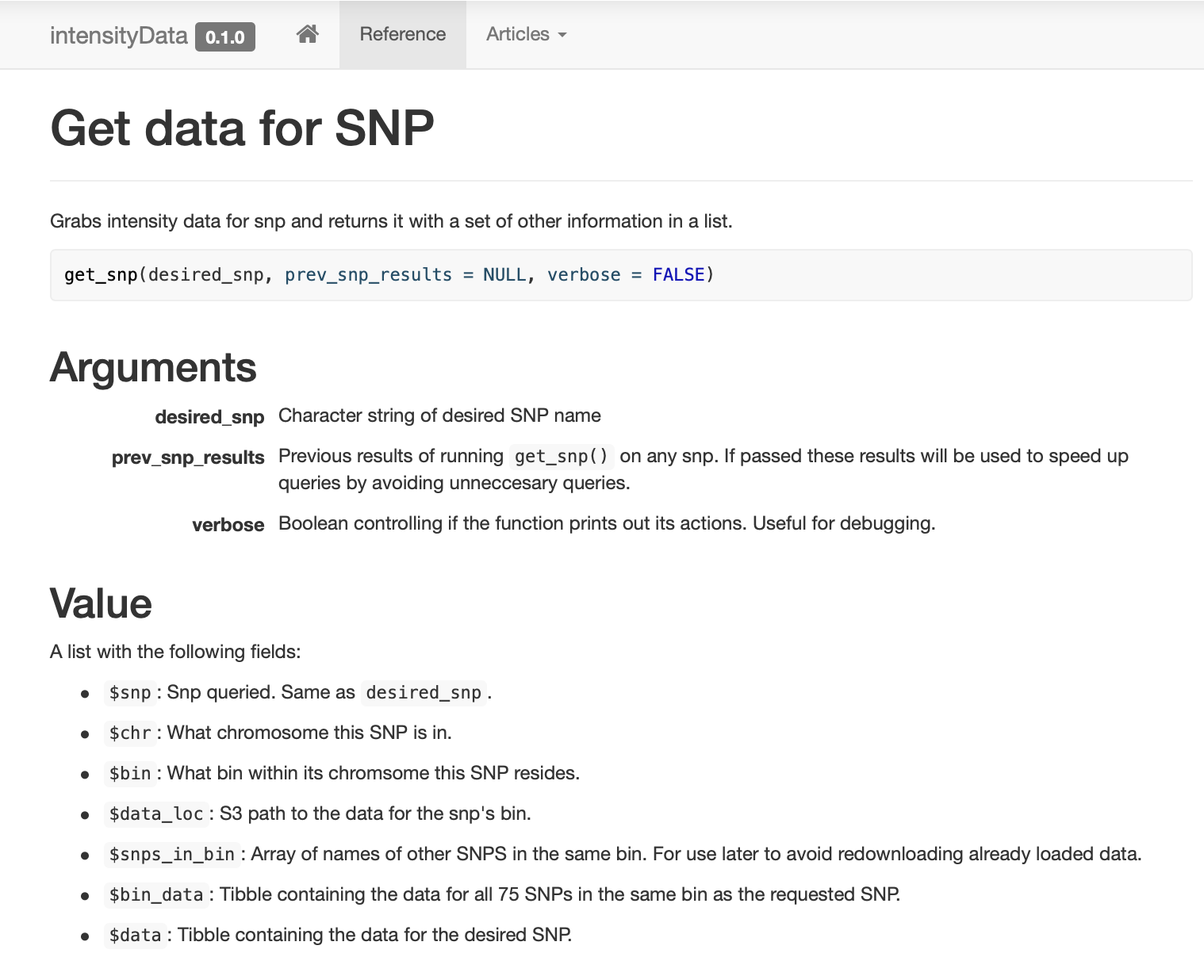
: , !
SNP , (binning) . SNP, (bin). ( ) .
, . , . ,
dplyr::filter , , .
,
prev_snp_results snps_in_bin . SNP (bin), , . SNP (bin) :
Resultados
( ) , . , . .
, , , …
. . ( ), , (bin) , SNP 0,1 , , S3 .
Conclusão
— . , . , . , , , . , , , , . , , , , - .
. , , «» , . .
:
- 25 ;
- Parquet- ;
- Spark ;
- 2,5 ;
- , Spark;
- ;
- Spark , ;
- , , - 1980-;
gnu parallel — , ;
- Spark ;
- Spark ;
- AWK ;
stdin stdout R-, ;
- S3 ;
- — ;
- , ;
- API ;
- , !