No
artigo detalhado anterior sobre o
Genoma Completo, prometemos publicar três problemas e testar o que resolver primeiro os três corretamente. Ao mesmo tempo, damos exemplos de como trabalhar com dados genéticos nessas tarefas. Hoje publicamos o primeiro.

No primeiro
artigo, compartilhamos informações e links úteis para trabalhar com dados de bioinformática. Recomendamos que você o leia primeiro, caso tenha perdido.
Isenção de responsabilidadeO trabalho com dados genéticos é realizado nos sistemas Unix (Linux, macOS), pois alguns comandos e software não estão disponíveis no Windows. Portanto, para usuários do Windows, uma das soluções mais simples é alugar uma máquina virtual Linux.
Todas as operações descritas abaixo são executadas na linha de comando - terminal. Antes de começar, aprenda a trabalhar em um terminal executando seu sistema operacional e use comandos, pois alguns deles podem prejudicar o sistema operacional e seus dados.
Software Necessário
Coletamos a
imagem de uma máquina virtual (VM) com todo o software necessário no Yandex.Cloud. Registre-se no Yandex.Cloud, na sua conta na seção Compute Cloud, clique em Criar VM. Como imagem pública, selecione 1000 genomas no catálogo Atlas Data Analysis.
Configuração da VM: 100% 2vCPU, 8GB RAM, 20GB HDD. Ao criar uma VM, você deve inserir os dados de pagamento, mas nada é baixado da conta. Uma partida e uma concessão adicional em uma palavra de código são suficientes para trabalhar com uma VM e uma imagem da Atlas até 31 de dezembro de 2019 gratuitamente. Para receber uma concessão para concluir tarefas, envie a palavra de código "ATLAS" para o
suporte do Yandex.Cloud .
Nota: a concessão é válida para novos usuários do Yandex.Cloud que abriram uma conta desde 18 de dezembro de 2019 ou para aqueles que ainda têm um período de avaliação e têm uma concessão inicial. A palavra de código ATLAS é válida apenas uma vez.
Primeiro, crie uma chave ssh no computador local a partir do qual você planeja se conectar à VM:
ssh-keygen -o -t rsa -b 4096 -C "my-local-machine" -f ~/.ssh/yandex-cloud -a 100
Não esqueça de copiar o conteúdo do
~/.ssh/yandex-cloud.pub para a janela apropriada ao criar a VM.
Se você deseja instalar o software no seu computador, abaixo estão todas as informações de instalação. Se você decidir usar o Yandex.Cloud, crie uma VM e prossiga para a próxima seção.
Plink
O Plink é um pacote de software para manipulação de dados genéticos e a pesquisa de associação de genoma amplo (GWAS). Foi desenvolvido pelo geneticista Sean Purcell (Shaun Purcell). Desde 2008, com a ajuda do Plink, centenas de GWASs são realizados em todo o mundo, cujos resultados são os melhores que a Atlas usa como fonte de dados para algoritmos de cálculo de riscos de doenças.
O Plink oferece um conjunto de ferramentas para armazenar e converter dados de genotipagem e pesquisá-los. O Plink também permite processamento estatístico, análise de desequilíbrio de ligação (LD), análise de identidade por descida (IBD) e identidade (estado por IBS), estratificação populacional e testes de epistasia - a interação de várias variações genéticas entre si.
O IBD e o IBS são usados para analisar a composição da população e determinar o parentesco.
Um exemplo de epistasia são as variações de rs7412 e rs429358 no gene APOE, uma certa combinação de variantes que aumenta acentuadamente o risco de desenvolver a doença de Alzheimer, enquanto cada variante individualmente contribui apenas com uma pequena contribuição para o risco.
Faça o download da versão estável do Plink no
site oficial.
BCFtools
BCFtools é um conjunto de utilitários para manipulação de dados genéticos no formato VCF e sua contraparte binária BCF. A lista de possíveis aplicativos do pacote BCFtools inclui anotação, filtragem, mesclagem e divisão de arquivos VCF / BCF, localização de interseções, indexação, pesquisa seletiva, classificação, contagem de estatísticas, etc.
Para instalar, faça:
git clone git://github.com/samtools/htslib.git git clone git://github.com/samtools/bcftools.git cd bcftools
O processo de instalação é descrito em mais detalhes
aqui .
KING
O pacote KING (INference for Gwas baseado em parentesco) é usado em estudos populacionais quando se trabalha com dados de uma pesquisa em todo o genoma de associações para determinar as relações de parentesco nos dados estudados. Nesta tarefa, o KING ajudará a determinar o grau de parentesco de várias amostras do projeto 1000 Genomes.
Você pode baixá-lo
aqui . Para resolver problemas, o manual do KING está disponível
aqui .
Quase todos os erros que podem surgir durante o trabalho com as ferramentas são descritos no Stackoverflow ou em seu equivalente bioinformático - Biostars .
Dados utilizados
Para orientação, usamos os dados abertos
do projeto 1000 Genomes. Para análise, selecionamos 10 amostras com informações sobre os genótipos de cerca de 85 milhões de variações obtidas pela análise de dados NGS alinhados com a versão do genoma de referência GRCh37. Os relacionamentos familiares e as populações de amostras são mostrados na Figura 1.
 Figura 1
Figura 1 Linhagem usada em amostras de VCF. O quadrado corresponde ao sexo masculino, o círculo à fêmea. Linha pontilhada significa parentesco indeterminado de segunda ordem.
Tome nota
O formato VCF permite armazenar informações sobre o campo de uma pessoa como um único número, se essas informações forem conhecidas durante a geração do VCF. É assim: o campo GT (genótipo, genótipo) para registros do cromossomo X contém um valor numérico que corresponde a um alelo, para homens e dois para mulheres. Se não houver informações sobre o campo biológico da amostra sequenciada, o campo GT conterá, por padrão, dois valores numéricos (destacados em vermelho na Figura 2).
Nos arquivos VCF usados neste manual, o cromossomo Y é excluído, mas a presença do cromossomo Y no arquivo VCF nem sempre significa que a amostra sequenciada realmente o possui. Isso ocorre devido às regiões pseudo-autossômicas (PARs), que são idênticas para os cromossomos X e Y e estão localizadas em suas extremidades.
Diferentes cromossomos normalmente não têm regiões longas e idênticas (homólogas), no entanto, os cromossomos X e Y possuem várias regiões com vários milhões de pares de bases no início (PAR1) e no final (PAR2). Portanto, ao analisar os dados de NGS em homens nas regiões PAR, são encontrados dois alelos (um para cada cromossomo sexual) e, nas mulheres, os genótipos podem aparecer nas regiões PAR do cromossomo Y, embora, na verdade, sejam genótipos do cromossomo X.
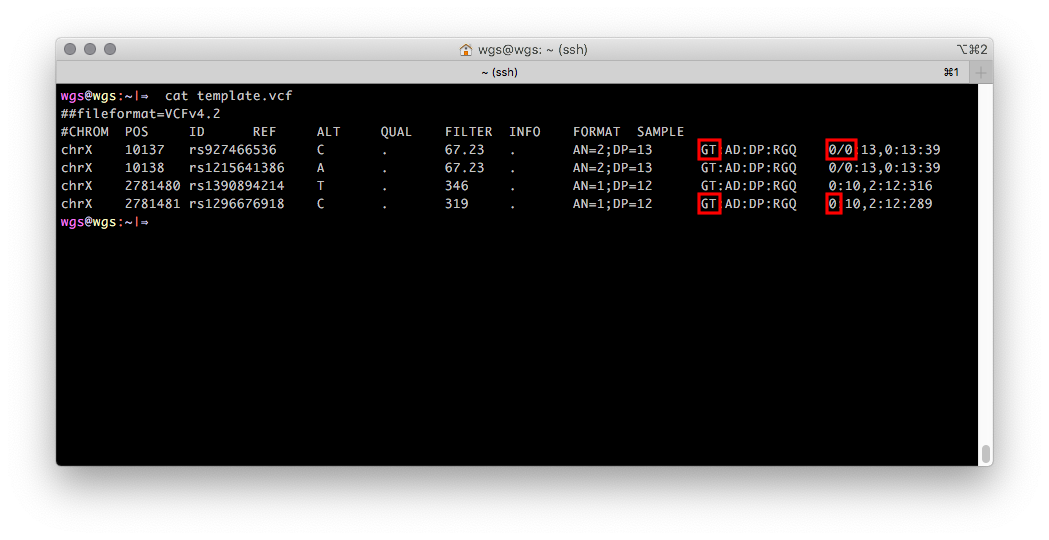 Figura 2
Figura 2 Arquivo VCF com genótipos do cromossomo X de um homem da região PAR1 (duas primeiras entradas) e região não pseudo-autossômica (duas últimas entradas).
Unidade educacional
O gênero genético é um conjunto de cromossomos sexuais que corresponde à manifestação das características sexuais primárias e secundárias de um tipo masculino ou feminino. Normalmente, os homens têm um cromossomo X e um cromossomo Y, enquanto as mulheres têm dois cromossomos X. Com vários distúrbios na formação de células germinativas, óvulos e espermatozóides, uma criança com um excelente conjunto de cromossomos sexuais pode nascer dos pais, o que geralmente leva a distúrbios do desenvolvimento características sexuais primárias e secundárias.
As duas anomalias sexuais cromossômicas mais comuns são a síndrome de Turner (um conjunto de cromossomos X0, ou seja, apenas um cromossomo X) e a síndrome de Klinefelter (um conjunto de cromossomos XXY).
Um alelo é um ou mais nucleotídeos que estão localizados em qualquer posição no genoma e têm uma alternativa. O conceito é usado para descrever genótipos. Distinga entre alelos de referência e alternativa. Todos eles são armazenados no arquivo VCF nos campos REF e ALT, respectivamente.
Determinar o sexo
Para usuários do Yandex.CloudTodos os dados para concluir as tarefas manuais e independentes são armazenados no Yandex.Cloud usando a estrutura mostrada abaixo. A pasta
Tutorial contém o arquivo VCF necessário para concluir o manual, a pasta
Test para tarefas independentes. A pasta
Technical contém dois arquivos com uma lista de identificadores de variações genéticas:
rsids_for_subsetting.txt usado no manual e tarefas para execução independente,
external_interpretation_rsids.txt pode ser necessário no futuro ao adquirir o seqüenciamento genômico no Atlas para fazer upload de dados de genotipagem para serviços de terceiros. A pasta
Tools contém, entre outras coisas, dois scripts usados nas tarefas 2 e 3.
home └── ubuntu ├── Data │ ├── Test │ │ ├── CEI.1kg.2019.test.vcf.gz │ │ └── CEI.1kg.2019.test.vcf.gz.tbi │ └── Tutorial │ ├── CEI.1kg.2019.demo.vcf.gz │ └── CEI.1kg.2019.demo.vcf.gz.tbi ├── Technical │ ├── external_interpretation_rsids.txt │ └── rsids_for_subsetting.txt └── Tools ├── convert_plink_delimiter.sh └── create_23andme.sh
Uma pasta será criada no diretório
/home na VM Yandex.Cloud, cujo nome corresponde ao nome de usuário especificado no estágio de criação da VM. Copie tudo do diretório
/home/ubuntu para o seu diretório através dos seguintes comandos:
cd ~ cp -r /home/ubuntu/* ./
Para o restoAo trabalhar em um PC pessoal, você pode baixar os arquivos necessários para a primeira tarefa no
link . O arquivo baixado suporta uma estrutura de armazenamento de arquivos semelhante à usada no Yandex.Cloud:
home └── ubuntu ├── Data │ ├── Test │ │ ├── CEI.1kg.2019.test.vcf.gz │ │ └── CEI.1kg.2019.test.vcf.gz.tbi │ └── Tutorial │ ├── CEI.1kg.2019.demo.vcf.gz │ └── CEI.1kg.2019.demo.vcf.gz.tbi ├── Technical │ ├── external_interpretation_rsids.txt │ └── rsids_for_subsetting.txt └── Tools ├── convert_plink_delimiter.sh └── create_23andme.sh
Descompacte o arquivo
atlas_wgs_contest.tar.gz com o comando
tar -xvzf atlas_wgs_contest.tar.gz Os arquivos VCF para executar tarefas de forma não arquivada ocupam cerca de 19 gigabytes cada, portanto, para economizar espaço, recomendamos trabalhar apenas com arquivos. Todos os programas listados acima já são capazes de trabalhar com dados de VCF compactados. Além disso, você não precisa fazer nada.
Para determinar o sexo do sujeito, é necessário examinar os genótipos no cromossomo X e excluir as regiões PAR1 e PAR2 localizadas no início e no final. Esses são os intervalos das posições 60001 a 2699520 e 154931044 a 155260560 na versão GRCh37 do genoma. Se o genótipo contiver uma designação numérica, esse é o sexo biológico masculino, caso contrário, a fêmea. Deve-se ter em mente que a designação de gênero no arquivo VCF depende da disponibilidade de informações sobre o campo biológico durante a geração do VCF, portanto, essa abordagem nem sempre pode ser usada.
Use o seguinte comando para cada uma das amostras no conjunto de dados. Substitua o identificador de amostra após o argumento
-s :
(/Data/Tutotrial/CEI.1kg.2019.demo.vcf.gz):
Ao executar os comandos, você verá uma parte do conteúdo do arquivo VCF para o identificador de amostra especificado. O
-r chrX:2699521-154931043 no BCFtools restringe a visualização do conteúdo do arquivo à região do cromossomo X da posição 2699521 para a posição 154931043 (região não PAR na Figura 3). Esses limites excluem regiões pseudo-autossômicas desnecessárias neste caso (PAR1 e PAR2). Usando os valores numéricos no campo GT, determine o sexo de cada amostra.
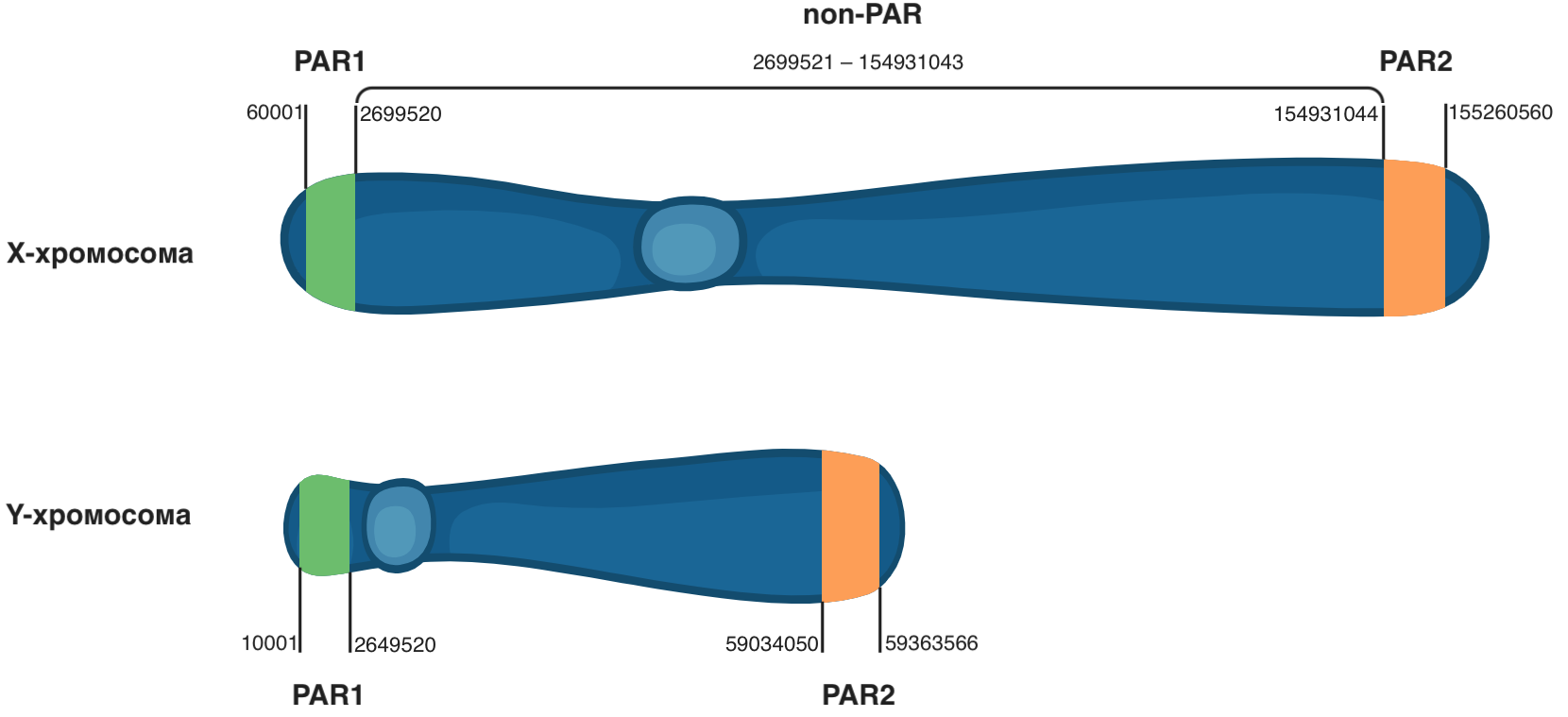 Figura 3
Figura 3 A localização das regiões pseudo-autossômicas de PAR1 e PAR2 nos cromossomos sexuais.
Você pode ver a lista de todos os identificadores de amostra no arquivo VCF na Figura 1 ou na última linha do cabeçalho do arquivo VCF. Eles serão listados após o nome da coluna FORMAT:
O verdadeiro sexo dessas amostras também é mostrado na Figura 1.
Nós determinamos o relacionamento
Para determinar a relação, precisamos comparar aos pares os dados genéticos de todas as amostras. É difícil fazer isso de acordo com o genoma completo: nesse caso, um arquivo VCF consome dezenas de gigabytes. O VCF que usamos ocupa apenas cerca de 2 gigabytes, mas ainda o filtramos de acordo com a lista de identificadores de variação genética (rsIDs) genotipados em chips da Illumina: GSA v1, GSA v2, HumanOmniExpress v1.0, HumanOmniExpress v1.3, InfiniumExome v1. 1 e Infinium OmniExpressExome v1.4. Estes são os chips mais populares na genotipagem comercial.
Nós compilamos uma lista de todos os identificadores de variações genéticas desses chips em um arquivo separado com uma lista de rsIDs. Ele contém 1,4 milhões de identificadores. Para filtrar o arquivo VCF, execute o seguinte comando:
bcftools view -O z -i 'ID=@rsids_for_subsetting.txt' CEI.1kg.2019.demo.vcf.gz > CEI.1kg.2019.demo.subset.vcf.gz
Cada vez que você usa o BCFtools e outros pacotes para trabalhar com arquivos VCF, o histórico dos comandos anteriores é adicionado ao cabeçalho do arquivo. Independentemente do método de filtragem do arquivo VCF e dos comandos executados anteriormente, você pode verificar a integridade e a identidade do conteúdo principal do VCF calculando a soma de hash:
O comando
gunzip -c descompacta o arquivo e
gunzip -c seu conteúdo em stdout, a partir do qual as linhas de cabeçalho do arquivo VCF iniciadas
# são excluídas (portanto, o comando
grep -v "^#" é usado). O cabeçalho é removido para comparar a integridade apenas dos dados genéticos, e não dos metadados sobre quais ferramentas e quando foram usadas para trabalhar com esse arquivo VCF.
Se o valor do hash corresponder, é possível seguir em frente e converter o VCF para o formato interno do Plink (por padrão, o formato do Plink é de três arquivos com as extensões bed, bim e fam). Nesses arquivos, apenas o genótipo, cromossomo, posição e alguns outros dados permanecem, e o restante é eliminado. Com esse formato, é muito mais fácil trabalhar e resolver vários problemas que não exigem informações adicionais do VCF. Por exemplo, conduza o GWAS.
Este comando criará três arquivos na pasta:
CEI.1kg.2019.demo.subset.bed
CEI.1kg.2019.demo.subset.bim
CEI.1kg.2019.demo.subset.famVocê pode determinar parentesco pareado para todas as 10 amostras. Usamos o seguinte comando para analisar arquivos Plink:
king -b CEI.1kg.2019.demo.subset.bed --kinship --prefix CEI.1kg.2019.demo.subset.kinship_analysis
Observe o arquivo
CEI.1kg.2019.demo.subset.kinship_analysis.kin0 e preste atenção à coluna Parentesco, que contém os coeficientes de parentesco para os pares de amostras indicados em ID1 e ID2, respectivamente.
Compare os coeficientes que você obteve no arquivo
CEI.1kg.2019.demo.subset.kinship_analysis.kin0 para todos os pares de amostras com o pedigree mostrado na Figura 1 (a linha tracejada corresponde ao parentesco de segunda ordem; no entanto, não há dados de parentesco exatos, ou seja, pode haver primos, tia / sobrinho ou tio / sobrinha). Tente fazer sua própria conclusão sobre quais valores dos coeficientes de parentesco podem corresponder à relação de primeira e segunda ordem.
SugestãoTrecho da documentação do KING: coeficientes de parentesco> 0,354 correspondem a amostras duplicadas ou gêmeos idênticos, de 0,177 a 0,354 a parentesco de primeira ordem (pais-filhos, irmãos), de 0,0884 a 0,177 a parentesco de segunda ordem (primos, tias / tios-sobrinhos) e de 0,0442 a 0,0884 - até o parentesco de terceira ordem (avós, netos, primos em segundo grau). Qualquer coisa menor que 0,0442 é difícil de interpretar sem ambiguidade.
A primeira tarefa da competição
Usando um conjunto de dados de teste de 12 amostras
Data/Test/CEI.1kg.2019.test.vcf.gz ,
Data/Test/CEI.1kg.2019.test.vcf.gz sua linhagem, guiada pelos resultados da determinação do sexo e análise de parentesco. Amostras que, de acordo com os resultados da análise, não estão em parentesco com alguém, escrevem nas proximidades, sem conectá-las a uma linha com outras amostras. O pedigree pode ser composto em um estilo semelhante à Figura 1, no entanto, isso permanece a seu critério. Os homens são indicados por um quadrado, as mulheres por um círculo, o casamento por uma linha horizontal, um filho por uma linha vertical, vários filhos por uma ramificação horizontal de uma linha vertical (na forma da letra P). Leia mais sobre essas designações
aqui .
Como escrevemos acima, os coeficientes de parentesco não podem caracterizar inequivocamente o parentesco de uma ou de outra ordem: os mesmos coeficientes de parentesco são obtidos ao comparar os pares pai-filho e irmão-irmã (parentesco de primeira ordem). Se não for possível estabelecer a natureza do relacionamento, indique algum dos possíveis. Observe que as amostras no conjunto de dados de teste têm identificadores diferentes daqueles usados no conjunto de dados de treinamento.
As respostas
devem ser enviadas para o correio
wgs@atlas.ru até 26 de dezembro às 23:59. Mais duas tarefas serão publicadas em breve, e os resultados finais das tarefas aparecerão em 28 de dezembro. O vencedor receberá o teste do Genoma Completo, e o segundo e o terceiro lugares receberão o teste genético do Atlas. Também haverá prêmios especiais do
Yandex.Cloud . Ex-funcionários atuais e atuais da Atlas não participam da competição;)