Continuamos o ciclo de tarefas, onde falamos sobre como trabalhar com dados genéticos. A primeira
tarefa “Descobrir gênero e grau de relacionamento” já pode ser resolvida e nos enviar respostas. Hoje publicamos o segundo.
O prêmio principal é o
Genoma Completo .

Anteriormente, compartilhamos informações e links úteis que podem ser úteis para trabalhar com dados de bioinformática. Recomendamos que você leia os artigos anteriores primeiro se os tiver perdido:
Qual é o genoma completo e por que é necessárioTarefa número 1. Descubra o sexo e o grau de relacionamento.Isenção de responsabilidade
O trabalho com dados genéticos é realizado nos sistemas Unix (Linux, macOS), pois alguns comandos e software não estão disponíveis no Windows. Portanto, para usuários do Windows, uma das soluções mais simples é alugar uma máquina virtual Linux.
Todas as operações descritas abaixo são executadas na linha de comando - terminal. Antes de começar, aprenda a trabalhar em um terminal executando seu sistema operacional e use comandos, pois alguns deles podem prejudicar o sistema operacional e seus dados.
Software Necessário
Coletamos a
imagem de uma máquina virtual (VM) com todo o software necessário no Yandex.Cloud. As instruções para configurar a VM e instalar o software podem ser encontradas no
artigo anterior, com a Tarefa nº 1.
Desta vez, você precisará construir um gráfico de dispersão bidimensional usando dados obtidos pelo método de análise dos componentes principais. Sugerimos construir esse diagrama usando qualquer software conveniente para você: Excel, Planilhas Google, Python, R e outros.
Para concluir a tarefa, você precisa do pacote de software Plink 1.9. Se você ainda não o instalou (e não concluiu a tarefa nº 1), leia o artigo anterior. Ele contém instruções de instalação. Para participar da competição de Ano Novo de 2019, todas as tarefas devem ser concluídas!
Tome nota
A análise de componentes principais (PCA) é um dos algoritmos de aprendizado de máquina sem professor quando uma máquina procura independentemente padrões nos dados. Na genética, o PCA permite agrupar amostras de acordo com dados de genotipagem em um espaço N-dimensional (geralmente bidimensional), onde os principais componentes obtidos explicam com mais precisão a variabilidade dos dados genéticos de amostra para amostra.
Ao conduzir essa análise, as amostras de uma população geralmente formam um aglomerado, cujo tamanho e suavidade dos limites dependem da semelhança das amostras em uma dada população. É provável que o algoritmo identifique amostras de diferentes populações em diferentes grupos. E amostras de populações próximas que pertencem à mesma superpopulação, por exemplo, EAS - Leste Asiático, como na Figura 1, serão identificadas próximas umas das outras ou mesmo em grupos que se cruzam.
 Figura 1
Figura 1 O pedigree das amostras utilizadas na VCF (o quadrado corresponde ao sexo masculino, o círculo à fêmea). A linha tracejada corresponde a um relacionamento indeterminado de segunda ordem.
Uma análise semelhante é usada para determinar a população por genotipagem. Para isso, é necessário um conjunto de dados de referência, que consiste em amostras com uma origem já conhecida. A conclusão sobre a população pode ser feita pelo agrupamento de amostras conhecidas mais próximo dos dados estudados.
Para simplificar, a essência da análise do PCA é que as distâncias entre pares no ponto multidimensional são conhecidas e esses pontos devem estar localizados em um espaço de menor dimensão, para que as novas distâncias entre pares sejam minimamente diferentes das originais. A redução de dimensão simplifica a análise de dados, mas quanto mais reduzimos, mais fortes as novas distâncias entre os pontos diferem da original. Portanto, a tarefa da análise de PCA também envolve encontrar um compromisso entre precisão e facilidade de análise. Tudo é como na vida.
A variante mais simples da execução de PCA em dados genéticos é baseada na identidade de alguns alelos, que podem ser divididos em dois subtipos: IBS (identidade por estado) e IBD (identidade por descendência). IBS significa a identidade de certos alelos em duas pessoas, mas não implica necessariamente o fato de qualquer relação entre eles. A IBD, ao contrário, fala da identidade de alelos devido à presença de um ancestral comum e, consequentemente, de parentesco.
Alelos IBD são inequivocamente alelos IBS, enquanto o inverso não é verdadeiro. No entanto, deve-se ter em mente que em algum momento viemos de um ancestral comum, portanto, alguns alelos podem ser IBD. Na análise PCA abaixo, apenas o conceito de IBS é usado, embora em análises mais complexas leve em consideração testes de significância estatística, limitações fenotípicas, tamanhos de cluster, idade e sexo da pessoa, além de informações adicionais sobre a estrutura da população.
Quanto maior o número de alelos diferentes em duas amostras, menos semelhantes e mais distantes um do outro. O valor do IBS para essas amostras será baixo. Mas para pais e filhos, o IBS será muito alto.
Conhecendo os valores do IBS para cada par de imagens no conjunto de dados, é possível executar uma análise PCA para ver como eles estão em cluster.
O teste genético Atlas utiliza um algoritmo muito mais sofisticado para determinar a representação populacional nos dados de genotipagem.
Dados utilizados
Lembramos que o manual usa dados abertos especialmente selecionados do projeto
1000 Genomes . Para análise, foram selecionadas 10 amostras com informações genotípicas de ~ 85 milhões de variações, as quais foram obtidas através da análise de dados NGS alinhados com a versão do genoma GRCh37. Os relacionamentos familiares e as populações dessas amostras são mostrados na Figura 1.
Construindo clusters de população
Use os três arquivos no formato Plink, obtidos anteriormente na Tarefa nº 1:
CEI.1kg.2019.demo.subset.bed CEI.1kg.2019.demo.subset.bim CEI.1kg.2019.demo.subset.fam
Determine a distância em pares entre todas as 10 amostras no conjunto de dados de treinamento e desenhe um PCA baseado no IBS (identidade por estado). Isso pode ser feito da seguinte maneira:
O parâmetro
—genome é apenas responsável pelo cálculo em pares de IBS / IBD entre todas as amostras no conjunto de dados. O parâmetro "
—read-genome " é a matriz de distância em pares obtida anteriormente, e os parâmetros "
—cluster —mds-plot 10 são responsáveis pela análise da PCA e pela saída de seus resultados para a tabela dos 10 primeiros componentes principais. De fato, essas são as coordenadas de cada amostra no espaço 10-dimensional.
O último comando criará 4 arquivos na pasta:
CEI.1kg.2019.demo.subset.clustering.cluster1 CEI.1kg.2019.demo.subset.clustering.cluster2 CEI.1kg.2019.demo.subset.clustering.cluster3 CEI.1kg.2019.demo.subset.clustering.mds
Vamos precisar dos dois últimos arquivos da lista.
A Figura 2 mostra como é o arquivo recebido no conjunto de dados de treinamento do MDS. Os campos FID (ID da família) e IID (ID individual) correspondem à família e aos identificadores de amostra individuais. Os campos C1 - C10 contêm os valores de cada um dos dez componentes principais de cada amostra, onde o componente C1 explica ao máximo a variabilidade dos dados genéticos das amostras analisadas e o C10 minimamente.
 Figura 2
Figura 2 Arquivo MDS com valores de 10 componentes principais para cada amostra.
Ao construir um diagrama de dispersão usando dois componentes (no espaço bidimensional), é possível detectar clusters correspondentes à população da amostra. A Figura 3 mostra gráficos de dispersão para os pares de componentes principais C1xC2, C2xC3 e C1xC3. Ao comparar os clusters obtidos com a afiliação populacional de referência (Figura 1), o par dos dois primeiros componentes C1 - C2 mostra a maior precisão (100%), separando corretamente todas as amostras de acordo com a afiliação populacional declarada no projeto 1000 Genomes. No entanto, sempre faz sentido comparar os resultados obtidos para vários pares de componentes devido à possível sobreposição ou separação de clusters reais.
 Figura 3
Figura 3 Gráficos de dispersão dos locais das amostras para pares de componentes principais; a localização dos marcadores foi ligeiramente alterada para evitar a sobreposição.
A criação de diagramas 3D usando os três primeiros componentes principais também pode ajudar a determinar o agrupamento, mas nem sempre. Por exemplo, a construção de um diagrama para os dados da Figura 3 nos permite identificar 4 grupos nos quais as amostras das populações PUR e KHV são agrupadas de acordo com a população, e as amostras da população CDX são divididas em dois grupos (Figura 4). Isso também é perceptível na Figura 3 nas coordenadas C2xC3 e C1xC3.
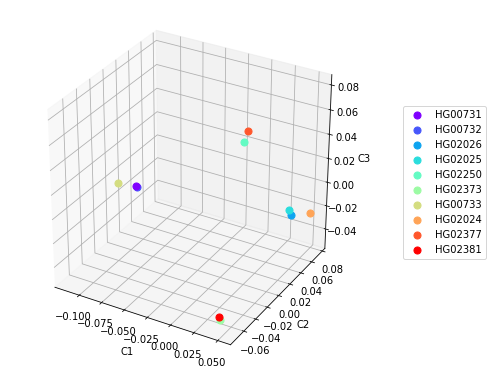 Figura 4
Figura 4 Gráficos de dispersão para os três componentes principais.
Tais resultados conflitantes da análise podem ser explicados por um pequeno número de amostras, uma vez que os valores dos componentes principais de cada amostra são diferentes para diferentes conjuntos de dados em tamanho e composição, e quando amostras adicionais de diferentes populações são incluídas, o resultado do agrupamento pode mudar. Os erros também são potencialmente possíveis ao criar um conjunto de dados e fornecer dados de referência sobre a população de amostras; no entanto, no projeto 1000 Genomes, a probabilidade de tal situação é bastante baixa.
Um arquivo MDS não usa tabulações ou vírgulas como delimitadores, portanto ajuste seu formato por conveniência. Use
tab ou
csv como o segundo argumento:
A equipe criará o arquivo
CEI.1kg.2019.demo.subset.clustering.mds.tab , no qual você pode baixar e criar gráficos de dispersão semelhantes aos mostrados na Figura 3. Compare os resultados, eles devem ser idênticos aos indicados acima.
Construindo uma árvore em cluster
Além disso, você pode avaliar o agrupamento de amostras usando uma árvore binária, que representa as informações de agrupamento sobre amostras de forma discreta. As informações sobre essa árvore estão contidas no arquivo
CEI.1kg.2019.demo.subset.clustering.cluster3 (Figura 5).
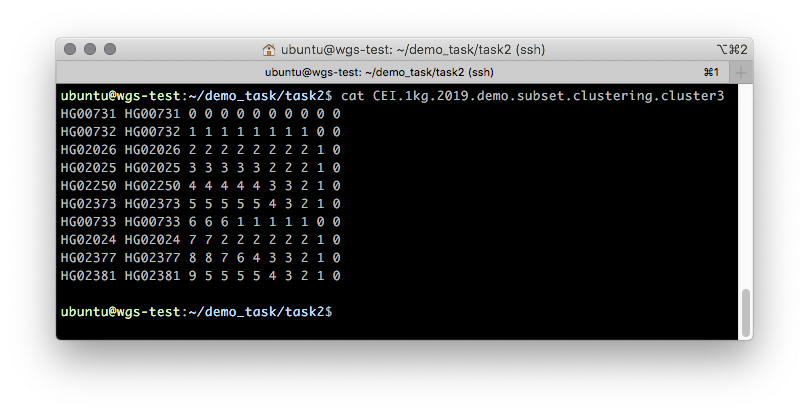 Figura 5
Figura 5 O conteúdo aproximado de um arquivo
.cluster3 que descreve o processo de agrupar em etapas as amostras de 1 cluster a N, em que N é o número de amostras.
As duas primeiras colunas deste arquivo contêm o FID e o IID. A afiliação de cluster é descrita por todos os outros. Este arquivo deve ser lido da direita para a esquerda nas colunas em incrementos de uma coluna: inicialmente, todas as amostras pertencem a um cluster “0” - a coluna mais à direita. Quando divididos em dois clusters (na segunda etapa, na segunda coluna), dois clusters aparecem: "0" e "1", onde o cluster "0" contém as amostras HG00731, HG00732 e HG00733 e o cluster "1" contém todo o restante. Uma ilustração dessa partição é mostrada na Figura 6.
Da árvore, pode-se concluir que as amostras pertencem à população (Figura 1). Além disso, a construção dessa árvore nos permite estabelecer a proximidade de populações individuais, ou seja, a ocorrência das populações CDX e KHV em uma superpopulação de EAS (já na primeira etapa de divisão das superpopulações, EAS e AMR são separadas em dois ramos existentes). Além disso, a construção de uma árvore em cluster pode ajudar a corrigir os resultados ambíguos da visualização de amostras nos componentes principais.
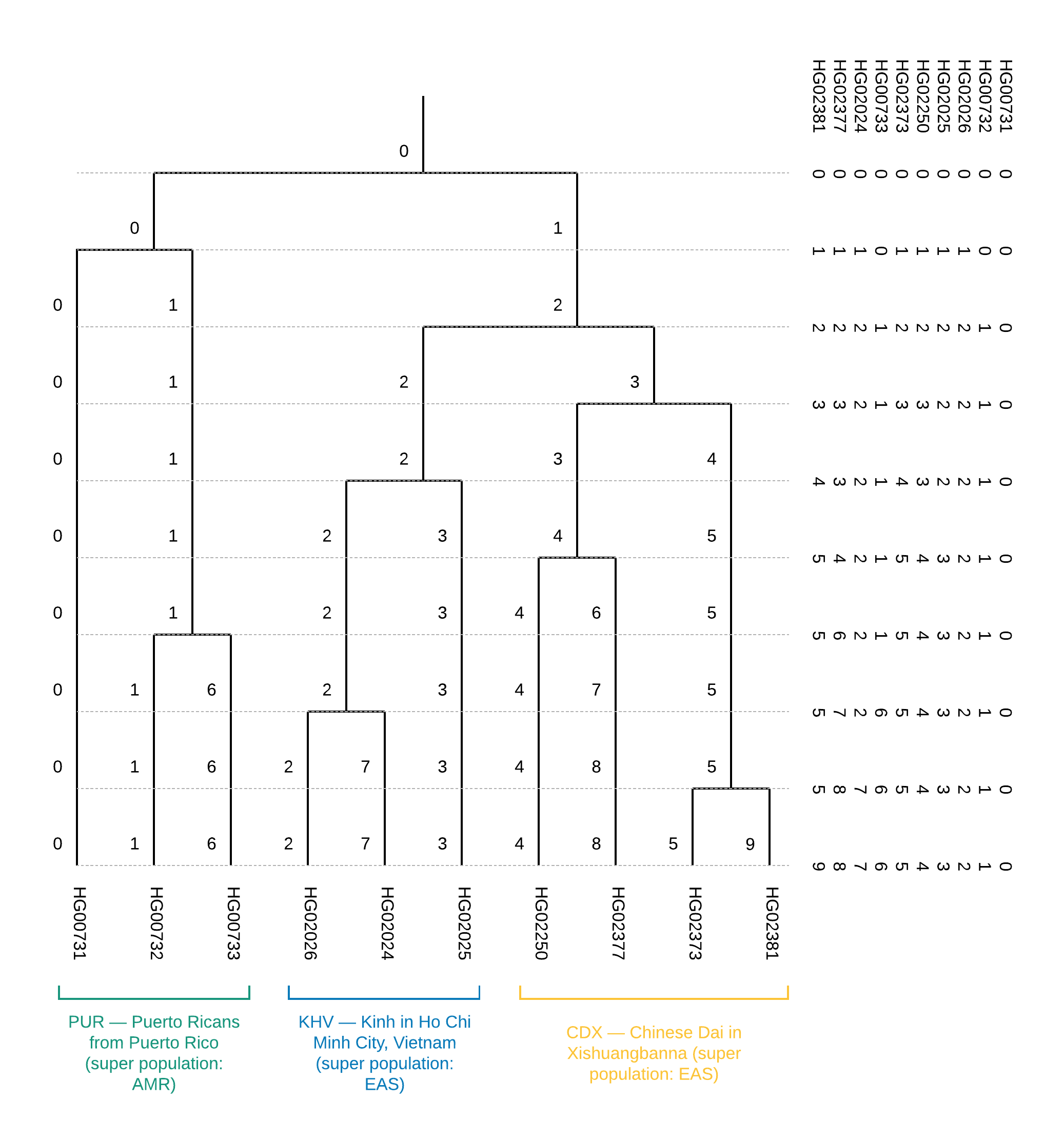 Figura 6
Figura 6 Árvore de
CEI.1kg.2019.demo.subset.clustering.cluster3 cluster binário para um conjunto de dados de treinamento de 10 amostras: à direita está o conteúdo do arquivo
CEI.1kg.2019.demo.subset.clustering.cluster3 (da direita para a esquerda no arquivo, identicamente de cima para baixo na figura).
A segunda tarefa da competição
Use o conjunto de dados de teste de 12 amostras de
Data/Test/CEI.1kg.2019.test.vcf.gz e o exemplo acima (Figura 5) para criar uma árvore de
.cluster3 cluster binário a partir do arquivo
.cluster3 que você
.cluster3 e anexá-lo à solução. Analise a árvore resultante e tire conclusões sobre o número de superpopulações apresentadas no conjunto de dados de teste.
Determine o agrupamento populacional de 12 amostras do conjunto de dados de teste analisando os principais componentes C1, C2 e C3, levando em consideração a árvore construída e indique-o no pedigree construído no Problema No. 1, restringindo os blocos populacionais individuais (semelhante à Figura 1). As amostras que não mostraram a presença de parentesco no Problema No. 1 devem ser colocadas da mesma maneira dentro dos blocos obtidos no diagrama sem conectá-los com linhas com outras amostras. Não se esqueça de anexar os gráficos de dispersão que você criou.
As respostas
devem ser enviadas para o correio
wgs@atlas.ru até 26 de dezembro às 23:59. Outra tarefa será publicada em breve e os resultados finais das tarefas aparecerão em 28 de dezembro. O vencedor receberá o teste do Genoma Completo, e o segundo e o terceiro lugares receberão o teste genético do Atlas. Também haverá prêmios especiais do
Yandex.Cloud . Ex-funcionários atuais e atuais da Atlas não participam da competição;)