Hoje publicamos a última tarefa do ciclo em que dizemos como trabalhar com dados genéticos.
As primeira e
segunda tarefas já foram publicadas: elas podem ser resolvidas e enviar respostas para nós. Avisamos que essa tarefa leva mais tempo que o resto.
O prêmio principal é o
Genoma Completo .

Anteriormente, compartilhamos informações e links úteis que podem ser úteis para trabalhar com dados de bioinformática. Recomendamos que você leia os artigos anteriores primeiro se os tiver perdido:
Qual é o genoma completo e por que é necessárioTarefa número 1. Descubra o sexo e o grau de relacionamento.Tarefa número 2. Determinação da estrutura populacionalIsenção de responsabilidadeO trabalho com dados genéticos é realizado nos sistemas Unix (Linux, macOS), pois alguns comandos e software não estão disponíveis no Windows. Portanto, para usuários do Windows, uma das soluções mais simples é alugar uma máquina virtual Linux.
Todas as operações descritas abaixo são executadas na linha de comando - terminal. Antes de começar, aprenda a trabalhar em um terminal executando seu sistema operacional e use comandos, pois alguns deles podem prejudicar o sistema operacional e seus dados.
Software Necessário
Coletamos a
imagem de uma máquina virtual (VM) com todo o software necessário no Yandex.Cloud. As instruções para configurar uma VM e instalar o software podem ser encontradas no
artigo com a primeira tarefa. Também há instruções sobre como configurar a máquina para usá-la gratuitamente até 31 de dezembro de 2019.
Nesta tarefa, você precisa converter os dados de genotipagem do formato VCF para o formato 23andMe, carregar os arquivos recebidos no serviço Promethease e familiarizar-se com o conteúdo do relatório para cada amostra.
O formato 23andMe é um formato de texto para armazenar dados de genotipagem e contém 4 campos separados por tabulações. O primeiro campo contém o identificador de variação (por exemplo, rsID), o segundo contém o cromossomo (valores válidos para esse campo são 1-22, X, Y e MT), o terceiro contém a posição no cromossomo, o quarto contém o genótipo (diplóide na presença de dois cromossomos homólogos, haplóides em outros casos). Esse formato é suportado por muitos serviços de interpretação; portanto, na tarefa trabalharemos com ele.
Para concluir a tarefa, você precisa do pacote de software BCFtools. Se você ainda não o instalou, leia o
artigo com a primeira tarefa. Ele contém instruções de instalação. Lembramos que para participar da competição de Ano Novo de 2019, todas as tarefas devem ser concluídas.
Além do BCFtools, você precisará
create_23andme.sh arquivo
create_23andme.sh - um script bash usado para gerar dados no formato 23andMe. Esse arquivo está localizado no diretório
/Technical no Yandex.Cloud, bem como no arquivo para download, disponível no link do
artigo .
Tome nota
Existem muitos serviços que analisam dados de genotipagem: MyHeritage, Promethease, FamilyTreeDNA, DNA.LAND, GEDmatch. Eles fornecem o download de dados de genotipagem em vários formatos, geralmente específicos para um provedor de genotipagem específico (Ancestry, 23andMe, MyHeritage, FamilyTreeDNA, GenesForGood e outros). O mais fiel ao formato dos dados é o Promethease: você pode baixar os arquivos VCF e 23andMe neste serviço.
Existem vários problemas de compatibilidade entre formatos e serviços:
- Empresas diferentes usam versões diferentes do genoma para mapear variações genéticas, o que é resolvido pelo chamado liftover, quando as posições das variações genéticas nos dados de origem são substituídas pelas correspondentes em outra versão do genoma. Por exemplo, o Atlas fornece dados de genotipagem para a versão do genoma GRCh38 e o GEDmatch recebe dados para a versão anterior do genoma GRCh37. A conversão das coordenadas das variações genéticas de GRCh38 para GRCh37 é chamada de elevador.
- Usando identificadores exclusivos para variações genéticas que não sejam rsIDs. Essas incompatibilidades são resolvidas excluindo essas entradas do arquivo ou anotando-as através da atribuição de um rsID. O segundo nem sempre é possível.
- Os serviços usam um conjunto fixo de variações genéticas. Às vezes, uma incompatibilidade de pelo menos parte dos dados baixados resulta em um erro de carregamento. Esse problema é relevante, por exemplo, para o MyHeritage. Isso pode ser resolvido destacando um conjunto de identificadores de variações genéticas que não causam erro de carregamento.
Dados utilizados
Lembramos que este manual utiliza dados abertos especialmente selecionados do projeto
1000 Genomes . Para análise, selecionamos 10 amostras com informações de genótipo de ~ 85 milhões de variações, que foram obtidas através da análise de dados NGS alinhados com a versão do genoma GRCh37. Os relacionamentos familiares e as populações dessas amostras são mostrados na Figura 1.
 Figura 1
Figura 1 O pedigree das amostras utilizadas na VCF (o quadrado corresponde ao sexo masculino, o círculo à fêmea). A linha tracejada corresponde a um relacionamento indeterminado de segunda ordem.
Conversão VCF
Abaixo estão as instruções para converter um arquivo VCF e carregar os dados recebidos no serviço Promethease, que recentemente se tornou gratuito. Sugerimos que você se familiarize com o relatório de Promethease recebido em qualquer uma das amostras. Use o arquivo VCF filtrado pela lista de variações obtidas na
tarefa nº 1 .
O comando
bcftools query permite extrair qualquer informação disponível em um formato especificado pelo usuário após o sinalizador
-f de um arquivo VCF. O sinalizador
-s indica o identificador da amostra (
HG00731 ) para a qual extrair dados. O sinalizador -e é usado para indicar critérios de exclusão, neste caso
'%ID=="."' Exclui entradas que não possuem um rsID. A saída da
bcftools query é passada para o script
create_23andme.sh , que converte os dados no formato TSV com 4 colunas (rsID, cromossomo, posição, genótipo) e os grava em um arquivo. Você pode fazer o download e salvar o script
create_23andme.sh para trabalhar com seus próprios dados de sequenciamento de genoma completo.
O script
create_23andme.sh usa os
create_23andme.sh extraídos do arquivo VCF para determinar o tipo de variação genética (variação de nucleotídeo único do SNV, inserção de INS ou exclusão de DEL) e escreve o identificador rsID, cromossomo, posição e alelos em
stdout acordo com o tipo específico de variação (A, G, T e C são alelos válidos para o tipo SNV, I e D são designações de alelos válidos para os tipos INS e DEL).
Lembre-se de que o processo de conversão leva muito tempo: cerca de 4 horas por arquivo para uma amostra com ~ 1 milhão de variações. Simultaneidade BCFtools não é suportado.
Vá para
promethease.com e registre-se. Clique no botão Carregar dados brutos (Figura 2) e faça upload do arquivo
HG00731.subset.23andme.txt . Após a conclusão do download, clique no botão Criar relatório gratuito e insira o nome desejado do relatório que será gerado de acordo com seus dados. Após a elaboração do relatório, você receberá uma notificação por e-mail e poderá se familiarizar com o conteúdo do relatório. Nos relatórios de cada amostra, localize o grupo sanguíneo determinado pelo sistema de interpretação da Promethease no sistema AB0 / Rh (fator Rh - Rh). Verifique seus resultados quanto à conformidade com a Tabela 1.
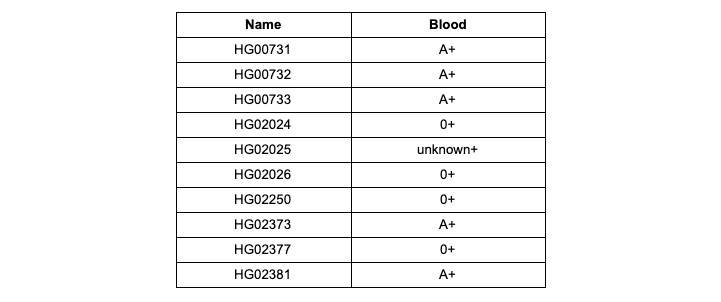 Quadro 1
Quadro 1 Grupos sanguíneos e fator Rh obtidos a partir de uma análise Promethease de amostras de um conjunto de dados demo
O atlas usa limites que diferem do Promethease para incluir um atributo específico na interpretação por nível de evidência. O nível de evidência refere-se à totalidade dos resultados de testes estatísticos e critérios para a significância de cada relação observada entre variação genética e qualquer característica do corpo humano. Muitas das características que podem ser encontradas no relatório de Promethease têm um baixo nível de evidência e / ou alto nível apenas em um conjunto limitado de populações, por exemplo, apenas para representantes da população asiática.
NotaEmpiricamente, instalamos uma lista de variações genéticas baseadas no chip
Infinium Global Screening Array v2.0 que pode ser carregado no MyHeritage. Esta lista (
external_interpretation_rsids.txt ) é armazenada em um arquivo separado no diretório
/Technical e pode ser usada para filtrar o VCF com a conversão subsequente por analogia com as instruções acima. Você também pode usar esse arquivo para filtrar dados de genotipagem de um chip, para que possa carregá-lo no MyHeritage. Se você já possui o teste genético Atlas, pode fazer o upload dos dados de genotipagem no formato da sua conta pessoal e filtrá-los de acordo com a lista de variações proposta - a primeira coluna nos dados carregados da sua conta pessoal.
Observe que os arquivos usados neste manual sempre contêm um campo ALT preenchido (alelo alternativo), o que possibilita entender a que tipo cada variação pertence (INS, DEL, SNV) e criar corretamente uma entrada no formato 23andMe. Os dados de seqüenciamento em todo o genoma no Atlas contêm o alelo ALT preenchido apenas nos locais onde esse alelo foi detectado; caso contrário, as informações para preencher o campo ALT ao gerar um arquivo VCF simplesmente não existem. A saída de dados em locais de referência homozigotos (posições no genoma em que o alelo de referência não foi encontrado) é necessária, pois não apenas as variações detectadas na sequência de nucleotídeos têm efeito clínico, mas também a ausência.
A ausência do alelo ALT nessas posições do genoma não nos permite determinar o tipo de variação genética para a qual apenas o alelo de referência (REF) foi encontrado. A gravação de genótipos para esses casos é complicada pela necessidade de usar uma fonte de informações sobre possíveis alelos para essa variação e não é coberta por este guia. Se você potencialmente usar este manual e o script
create_23andme.sh para converter um arquivo VCF obtido após um seqüenciamento em todo o genoma no Atlas, o arquivo convertido não conterá genótipos homozigotos de referência, pois o script
create_23andme.sh filtra explicitamente esses registros para eliminar erros ao criar registros para inserções e exclusões.
Para que o script
create_23andme.sh ainda produza genótipos homozigotos de referência, é necessário substituir o conteúdo das linhas 25 a 28 nele
... if [ "$ALT" == "." ] || [[ "$ALT" == *"*"* ]] then continue fi ...
em
... if [[ "$ALT" == *"*"* ]] then continue fi if [ "$ALT" == "." ] then echo -e "$RSID\t$CHR\t$POS\t$REF$REF" fi ...
Essa substituição permite que entradas
stdout com genótipos de referência homozigotos sejam exibidas. Deve-se ter em mente que essas entradas para inserções e exclusões estarão incorretas, pois os alelos válidos no formato usado para inserções e exclusões são I e D, e o script usará os alelos A, G, T ou C. Para gerar dados corretamente para inserções e deleções, é necessário conhecer antecipadamente que tipo de variação é característica de uma determinada posição do genoma em que o alelo ALT não foi detectado. Essas informações podem ser obtidas analisando o alelo ALT, se disponível (já implementado em
create_23andme.sh ) ou usando um banco de dados externo, por exemplo, dbSNP (não em
create_23andme.sh ).
Para obter um relatório do Promethease sobre um arquivo VCF completo do seqüenciamento genômico completo no Atlas, você pode fazer o upload do próprio arquivo VCF para o Promethease, no entanto, lembre-se de que o tamanho do arquivo compactado do Atlas VCF é de cerca de 8 gigabytes, enquanto o Promethease permite o upload de arquivos não mais que 4 gigabytes. A descrição das soluções para esse problema está disponível
aqui . Outra solução é dividir o arquivo VCF em várias partes (menos de 4 gigabytes cada) e carregar cada uma como um arquivo adicional no menu de download de dados do Promethease.
A terceira tarefa da competição
Faça o download dos dados convertidos de cada uma das 12 amostras do conjunto de dados de teste que você filtrou de acordo com a lista de variações da primeira tarefa, no Promethease e compile uma tabela de correspondência para o identificador da amostra - grupo sanguíneo AB0 / Rh definido pelo sistema de interpretação do Promethease (fator Rh - Rh). Grupos sanguíneos identificados probabilisticamente e registrados com o prefixo “prob” no relatório Promethease, escrevem sem o prefixo. Registre valores indefinidos como desconhecidos (o fator Rhesus para grupos sanguíneos desconhecidos ainda precisa ser gravado, se definido). Um exemplo é apresentado na Tabela 1.
A conversão do VCF para o formato usado acima na implementação proposta é bastante simplificada, mas requer uma quantidade significativa de tempo. Para otimização, você pode escrever um script com um loop que gerará esses dados automaticamente, repetindo um conjunto de identificadores. É possível criar vários scripts e para cada transferência conjuntos diferentes de identificadores de amostra para execução paralela; no entanto, o número de scripts em execução paralelo não deve exceder o número de CPU do seu computador / máquina virtual. Uma boa descrição da criação de tais loops está disponível
aqui . Ao trabalhar no Yandex.Cloud, você pode, se necessário, criar outra máquina virtual com um grande número de CPUs virtuais, o que reduzirá proporcionalmente o tempo necessário para concluir uma tarefa.
Esta é a última tarefa do nosso ciclo. As respostas
devem ser enviadas para o correio
wgs@atlas.ru até 26 de dezembro às 23:59. Publicaremos as respostas corretas e os nomes dos vencedores em 28 de dezembro. O vencedor receberá o teste do Genoma Completo, e o segundo e o terceiro lugares receberão o teste genético do Atlas. Também haverá prêmios especiais do
Yandex.Cloud . Ex-funcionários atuais e atuais da Atlas não participam da competição;)