根据我们的经验,使用DNA分析人口的归属关系会在公众中引起三个大问题:基因和族裔群体可以联系在一起吗?如何从技术角度分析起源,以及基因检测是否可以“鉴定犹太人”。 出于某种原因,正是具有DNA的犹太人身份问题使那些拥有不可否认的证据证明属于上帝拣选的人们以及那些不吃未发酵的食物并且不阅读律法书的人们感到担忧。
在Geektimes的新Genotek材料中,我们将尝试按顺序回答所有问题。 是的,我们还定义了犹太人。

在生物学,医学和遗传学领域又名种族群体
人类有一种不良习惯,即以一种种族相对于另一种种族的“先天”优势为暴力辩护-这就是为什么现代生物学家谨慎对待种群之间遗传差异的问题。 在整个20世纪,种族和族裔之间生物边界的(不存在)存在激烈的讨论,但是在这个问题上尚未达成最终共识(
1 )。
希望对人类基因组进行测序可以使所有人聚在一起。 从“从”到“到”的基因组将表明,群体之间的界线本质上是社会性的,每个人的基因都相同。 结果却不同:对人类核苷酸代码的仔细研究复活了,并且对种族和族裔群体之间生物学差异的兴趣增加了。 通常,相同的基因发现与疾病风险(
2 ),药物代谢(
3 ),人体对环境条件的反应(
4 )相关的等位基因变异略有不同,并且这些变异在不同的人群中以不同的频率发现。
已经停止了对不存在的“印度”或“非洲”基因的搜索,但是医学和人口遗传学领域的研究仍在参与者的生物学特征和种族之间进行了比较。 在此类作品中,“种族”和“民族”一词的使用得到了积极的讨论(并经常受到谴责)。 人们试图引入规则,迫使研究人员证明使用“滑溜”类别的必要性,并澄清特定术语的确切含义。 去年2月,最受人尊敬的科学期刊之一《科学》发表了一篇模棱两可的文章(
5 ),提议完全放弃在基因研究中使用“种族”一词,取而代之的是更正确和中立的“血统”-“起源” 。
但是即使在条件不确定的情况下,人类仍然可以分为人群:特别是为了正确进行药物的临床试验和评估疾病的风险。 例如,NOD2基因的三个等位基因变体-R702W,G908R和1007fs-与欧洲人患克罗恩氏病的风险增加有关(
6,7 ),然而,这些变体中没有一个与日语中的克罗恩氏病相关(
8 )。 已知CCR5基因的等位基因会影响HIV感染患者的免疫缺陷发展速度(
9 ):在其中发现了一种选择,可以减缓欧洲裔美国人的疾病发展速度,但可以加快其在非洲裔美国人中的发展速度(
10 )。 亚洲人发现,p53蛋白基因的多态性与调节人口的应激反应并抑制肿瘤的发展有关,与人口居住地的冬季平均温度之间存在相关性-基因适应霜冻(
11 )。 而且,如果过去只使用参与者自己提供的信息将样本分解成种族,那么在后基因组时代,他们会通过对受试者起源的遗传评估来对其进行补充和完善。
种群之间的遗传变异
在日常生活中,我们根据外貌或交流语言将人们分为几类。 大多数丹麦人彼此相似,而彼此之间却不像意大利人(
这是一个很酷的可视化效果,具有不同国籍的平均肖像)。 丹麦人和意大利人彼此之间的距离要远得多-距离撒哈拉以南非洲的居民更近:人类的表型根据地理模式聚集在一起。 基因型的分布具有相似的结构:通常,本地群体的成员比偏远地区的居民具有更紧密的家庭联系,居住在一个地区的人口比其栖息地被地理障碍(例如山脉或水域)隔开的人口更近数组)。
而且,人类的遗传多样性低于许多生物物种的遗传多样性。 人类是一个年轻的物种,这一事实可以解释这一点:各个群体只有很少的时间来积累差异。 两个随机选择的人彼此相差〜1000个核苷酸,而两个黑猩猩在〜500个“字母”中一次也不重合。 但是,人类基因组总共有大约300万个潜在的“差异点”。 这些差异中的大多数被称为单核苷酸多态性(SNP),是中性的或几乎中性的,但其中一些是造成人与人之间表型差异的原因。
世界人口中的中性多态性分布(由于它们没有生物学意义,因此没有经过定向进化选择,它们是由迁徙之风携带的)反映了我们物种的人口历史。 遗传和考古学证据表明,在过去的100,000年中,人口规模显着增长。 人们在非洲以外定居,殖民了世界其他地区。 安置过程以两种方式影响等位基因的地理分布:首先,“奠基者效应”受到影响-通常,在移民人口中,祖先人口的整个多样性库中仅代表了一部分遗传变异。 其次,发生了所谓的“分类穿越”。 对主要在其群体内形成,这限制了居住在不同地理区域的个体之间现有和新兴的从头多态性的分布。 这些过程导致遗传差异的逐步积累。
在人口群体的背景下,基因组标记在70年代至80年代开始研究,在90年代开始用于识别特定人群的基因。 研究人员反复证明,遗传多态性可以成功地隔离人群,并确定一个人的团体隶属关系。 然后表明,生活在同一大陆上的人们在基因上通常比来自不同大陆的人彼此更接近。 最初,在此类研究中,从一开始就了解有关出生地,种族,族裔的信息,并与遗传数据结合使用。 如果仅根据遗传特征盲目分配受试者,那么地理起源,种族和人口结构之间的对应关系就不太明显。 如进一步的研究显示,成功取决于所使用的遗传标记及其数量(越多越好),参考人群的正确选择和其他因素(
12 )。
到2004年,在美国,人口的遗传定义不仅被用于生物医学研究,而且还用于犯罪调查:《自然》杂志的
这篇文章包含了一个令人振奋的故事,讲述了警务人员如何拼命寻找罪犯,并命令一家商业公司进行DNA测试,犯罪嫌疑人肤色开了案。 关于遗传起源分析的建议成功地引起了人们过去对人们的普遍关注。 “根源躁狂症”在《时代》杂志的一篇文章中被称为“嗜好”,专门研究“美国的最新痴迷”-家谱研究。
研究人类起源和进化的专家们积极使用基因组方法。 例如,2013年,一个国际研究人员团队使用遗传分析来驳斥了来自卡扎尔人的阿什肯纳兹犹太人起源的假说(
13 )。 作者使用的基因组数据集是在公共领域:它包含100多个世界人口。 我们建议与我们一起进行一次小型研究:确定样本中Genotek客户的位置,同时了解确定人口的技术细节。
研究目的
在参考人群中识别Genotek客户。 找出我们的样本中是否有阿什肯纳兹犹太人的代表。 演示分析个体的原理和方法。
研究目标
使用ADMIXTURE程序,使用Behar等人,2013年的数据集作为训练样本,处理722名受试者的基因分型数据。
材料与方法
Behar等人(2013年)的原始工作使用了1,774人的数据:其中包括88个非犹太人口的代表(来自阿拉伯,中亚,东亚,欧洲,中东,北非,西伯利亚,南亚和亚美尼亚撒哈拉以南非洲地区)和18个犹太人口。 作者需要一个广泛的数据集来准确确定世界人口背景下的ashkenazes位置:任务是介绍该组可能来自的三个地理区域-欧洲,中东和Khazar Khaganate。 作者强调了代表现代欧洲,中东和犹太人口(即祖先人口的直接后代)的抽样方法与对应于Khazar Kaganate(大约在1000年前就不存在)的样本之间的区别。 问题是,没有一个人口是卡加尼人的直接继承人。 作为哈萨克人的现代代表,作者选择了南高加索地区(阿布哈兹人,亚美尼亚人,阿塞拜疆人,格鲁吉亚人),北高加索地区(阿迪格斯,巴尔卡尔斯,车臣人,卡巴丁人,奥塞梯人和其他几个民族),楚瓦什人和Ta人。
我们将来自俄罗斯各个地区的722人的样本添加到了数据集中。
为了进行统计分析,我们使用了ADMIXTURE程序,该程序使我们能够根据基因型数据估计一个人最可能的起源。 除此之外,所讨论文章的作者还使用了其他统计方法,对所提出的问题给出了类似的答案。 我们将专注于ADMIXTURE,因为正是这种算法使我们能够估计祖先群体对所研究基因组的百分比贡献。
ADMIXTURE在Markov链(Markov链Monte Carlo,MCMC)中使用蒙特卡洛方法。 这是该算法作者针对想要更详细地了解过程数学方面内容的人员的文章
链接 。
让我们看看ADMIXTURE如何处理我们集合中的样本和总体示例
总共,我们有2496个样本/个人,每个样本属于106个现代人口之一。 我们建议现代人口最有可能来自相对较少的祖先人口。 该分析中的“祖先种群”是一些古老的基因组簇,它们是根据遗传相似性原理联合在一起的。 ADMIXTURE既可以任意提出关于样本中此类簇数的假设,也可以选择最正确地描述基因组数据真实分布的最佳数。
收到有关基因型和“祖先”种群估计数(K)的信息后,ADMIXTURE建立了一个模型,用于估计每个“祖先”种群对每个样本的贡献。 在解释数据时,基因组的定量组成(簇的百分比)和定性的组分都非常重要-它们在特定基因组中的存在与否。 根据这些数据,可以对人口的进化过程做出假设,尤其是对人口群体中存在或不存在共同“根”的假设。 但是,如果我们构建的模型良好,则结论将是合理的:选择K.的最佳值。
我们选择K的最佳值
如何确定给定样本中最接近真实人口的“祖先”人口数量? 根据经验!
ADMIXTURE是一个聪明的程序:对于给定的数量K,基于个体基因型的数据(评估每个古代基因组对每个样本基因组的贡献)建立人口遗传结构模型,她不会忘记最后与现实进行比较。 检查构造模型对输入的描述程度。 比较的量度是“错误”-描述模型与实际数据之间不匹配的值。 误差越大,祖传人口数量的假设就越差。
如何选择K的最佳值? 我们在该样本上启动ADMIXTURE算法,替换为不同的K值,并为每个K获得自己的误差值。 我们绘制了误差大小对K的依赖关系。这是文章作者获得的图形:
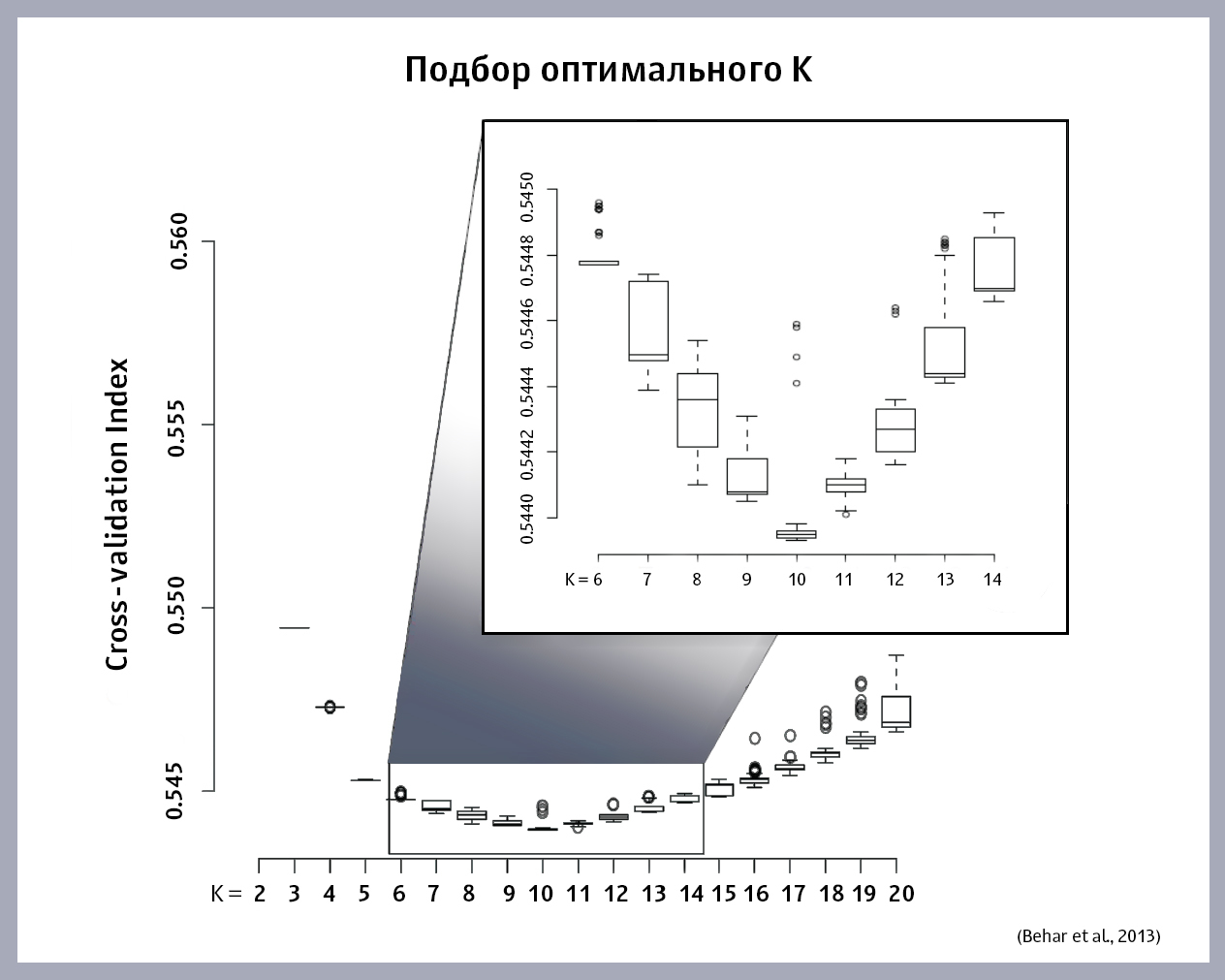
K的最佳值在函数的最小值。 如果在图形上未找到最小值(函数在不断增加或减小),则必须通过选择新的K来构建模型,直到找到正确的K。
即使选择了最佳的K,分析结果的可靠性也取决于样品的正确性:
1.个人之间不应有亲戚关系。
2.用于基因分型的单核苷酸多态性(SNP)应该以足够高的密度均匀地分布在基因组上。
3. SNP等位基因必须处于平衡连锁状态,也就是说,特定个体中给定等位基因存在的概率应仅取决于人群中该等位基因的频率,而不取决于基因组中的其他等位基因。
从图中可以看出,该样本的最佳K为10个“祖先”种群。
结果
这样的分析结果可以通过ADMIXTURE可视化(在该图中,只有一部分数据可见):
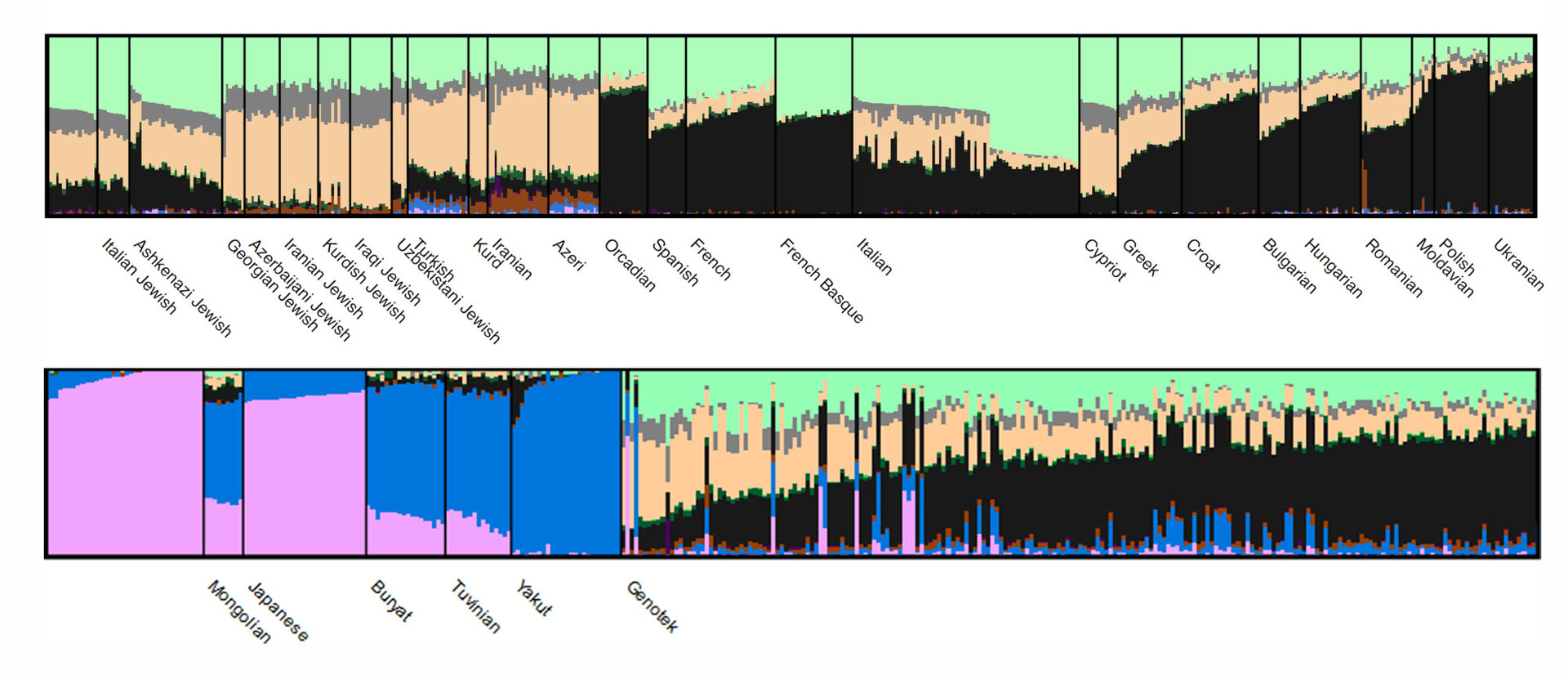
每个簇具有其自身的颜色,并且群体在基因组中簇的份额不同(或没有不同)。
这是图片的交互式版本,用于详细研究:移动鼠标并滚动以查看所有人群或更详细地考虑某些人群。
通常,在Genotek的“人口”中,聚类比率预计将对应于东欧血统的人口的特征模式。 有趣的是从单个样本的级别开始:
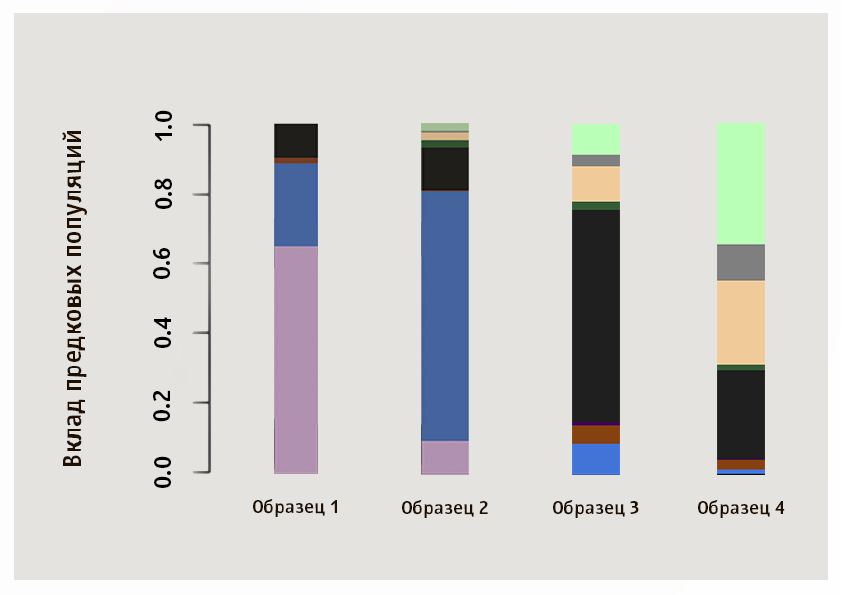
尽管最接近给定样本的总体由数值确定,但也可以通过视觉比较图案来获得很多信息。 我们建议您从图像中独立确定四个Genotek客户的样本的最近种群。
答案在这张照片中,样本1和2来自亚洲:粉红色的簇群在我们的样本中对于日本人和可汗人来说是典型的,雅库特人则是蓝色,第三个样本显示了俄罗斯人,白俄罗斯人,乌克兰人和波兰人所特有的成分比例,第四个是典型的Ashkenaz犹太人。
在722个样本中,我们找到了9个Ashkenazi犹太人。
结论
人口隶属关系远非决定一个人的种族自我认同的唯一因素。 但是,仍然有可能揭示族群与其代表的基因组结构之间的相关性。 这种分析既可用于科学和医学目的,也可用于所有研究者对其自身根源的研究。 同时,重要的是要了解模型正在不断改进,并且应结合其他数据(例如家谱树)考虑获得更高准确性的结果。
原始文章的作者未找到任何证据表明阿什肯纳兹的卡扎尔人起源。 基因测试当然会“知道”如何识别犹太人-但是,人们不应忘记“犹太人”首先是一种心理状态。
在不久的将来,更新的家谱DNA测试以及扩展的结果将在Genotek推出:我们将把人口数量增加到数百,增加犹太人口。 我们将为通过我们遗传材料的每个人更新您个人帐户中的信息。 如果您仍然没有基因型,我们邀请您
加入 。
参考文献
- Foster M.,Sharp R.(2002)。 种族,种族和基因组学:作为生物异质性的代理的社会分类。 基因组研究。
- Collins FS,McKusick VA(2001)。 人类基因组计划对医学的意义。 贾玛
- Nebert DW,Menon AG(2001)药物基因组学,种族和易感性基因。 药物基因组学
- Olden K.,Guthrie J.(2001)。 基因组学:对毒理学的影响。 笨蛋 Res。
- Yudell M.,Roberts D.,DeSalle R.,Tishkoff S.(2016年)。 从人类遗传学中淘汰种族。 科学。
- Ogura,Y。等。 (2001)。 NOD2的移码突变与克罗恩氏病的易感性有关。 自然。
- Hugot,JP等。 (2001)。 富含NOD2亮氨酸的重复变异与克罗恩氏病的易感性相关。 自然。
- Inoue,N.(2002)。 日本克罗恩氏病患者缺乏常见的NOD2变异体。 肠胃病学。
- Martin,MP等人(1998)。 CCR5启动子变体可加速AIDS进展的遗传过程。 科学。
- Gonzalez,E。等人(1999)。 与CCR5单倍型相关的种族特异性HIV-1疾病缓解作用。 程序 Natl Acad。 科学 美国
- 施宏等。 (2009)。冬季的温度和紫外线与东亚p53肿瘤抑制途径中的遗传变化紧密相关。美国人类遗传学杂志。
- Bamshad M.,Wooding S.,Salisbury B.等。(2004)。解构遗传学和种族之间的关系。Nat Rev Genet。
- Behar DM等。(2013)。从阿什肯纳兹犹太人的Khazar起源的全基因组数据中看不出任何证据。人类生物学