线粒体-小工人还是大老板?如果您认为我们共同生活的最重要故事是在婚礼期间开始的,那么事实并非如此。 每个人一生中最重要的故事始于十亿多年前,当时我们遥远的单细胞祖先被迫与我们现在称为线粒体的人签订“婚姻合同”(请参见共生理论)。
线粒体有两个膜(内部和外部)和它们自己的呈DNA形式的遗传物质(图1)。 线粒体内膜上有一个氧化磷酸化系统,该系统的运作提供了能量底物的氧化作用,并形成了ATP。
图 1 。 线粒体的示意图结构
在细胞和线粒体的婚姻契约中,没有“在疾病和健康方面”和“好”的条款。 如果线粒体变老,细胞可以在线粒体吞噬过程中杀死它,而线粒体又可以调节功能异常和陈旧细胞的凋亡过程。 如果相互质量控制的过程受到干扰,则会启动老化机制。 细胞凋亡的机制被破坏,不受线粒体控制的自由基数量增加。 这会引起全身性炎症,破坏细胞的DNA。 因此,MX功能障碍,与年龄有关的疾病,衰老和代谢功能障碍之间有很强的关系[1]。 代谢功能障碍是衰老的启示。
“像车轮上的松鼠”-线粒体的动力学
并非所有的代谢紊乱都归咎于我们的暴饮暴食。 代谢紊乱主要与线粒体无法应对营养有关。 细胞中的线粒体并不容易。 我们“喂食”我们的细胞太多或太少,然后向他们提出“要求”,以ATP的形式释放能量,ATP的数量必须完全符合我们的需求。 为了定期“摆脱”这种情况,线粒体确实使用了一些“运动”-分裂(裂变)和融合(融合)。 这些“线粒体运动”被统一为“线粒体动力学”。 线粒体分裂与融合之间的平衡是生物能适应细胞代谢需要的主要机制[2,3]。
大多数线粒体存在于能量需求很高的组织中,例如肌肉,肝脏,棕色脂肪组织和大脑。 不足为奇的是,这些组织中的线粒体动力学得到了更好的研究。
因此,如果这些组织中任何一个的细胞(除了大脑中的某些神经元,以后都会更多)吸收大量营养(摄入量超过成本),则线粒体处于分裂(碎片化)状态。 如果细胞处于饥饿状态(收入少于成本),则线粒体会合并并且处于连接状态。 [3,4]。 这就是维持细胞动态平衡的方式(图2)。
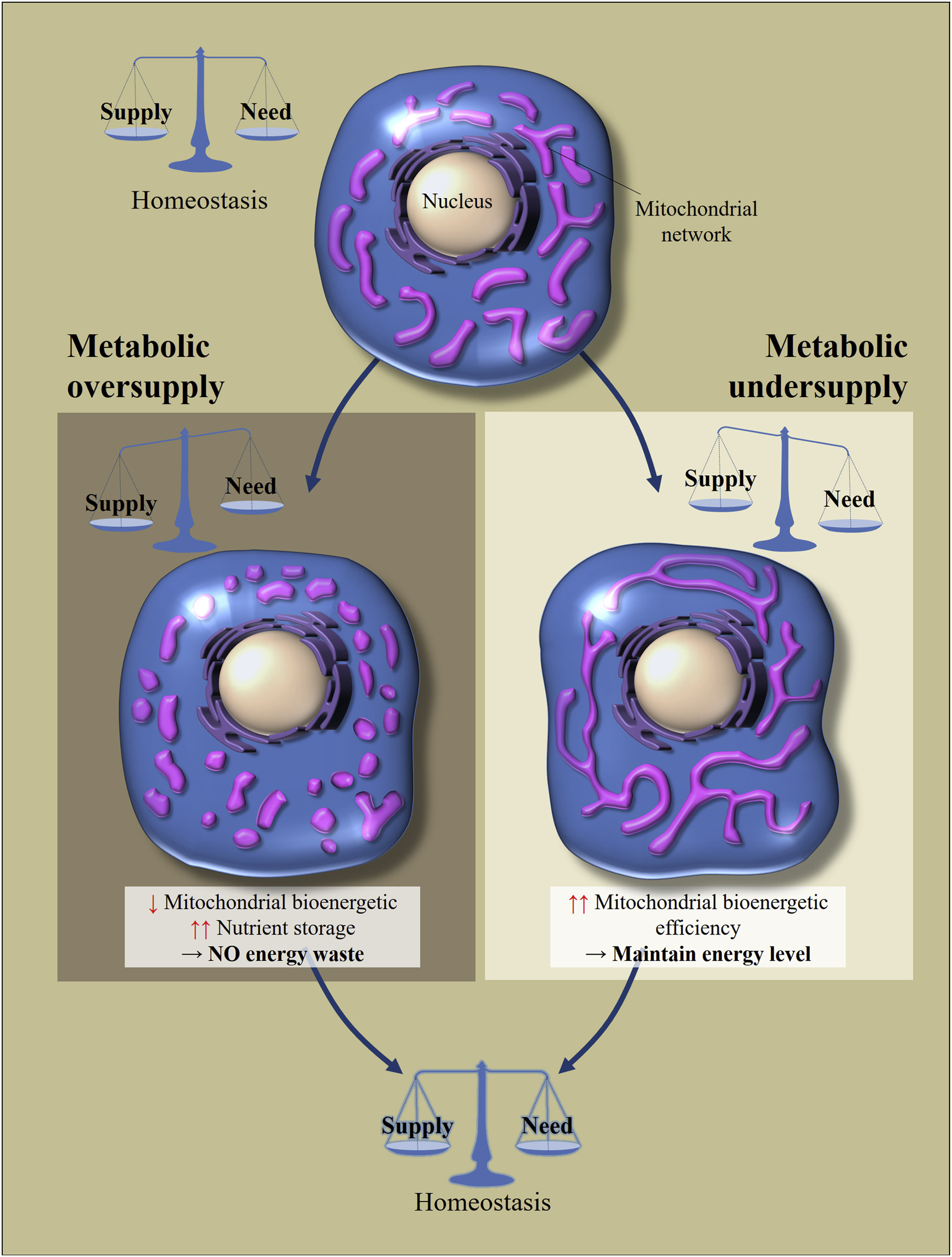 图 2
图 2调节线粒体的形态和生物能效率,以应对营养素摄入过多或不足[2分]
细胞代谢稳态取决于养分摄入与其消耗之间的平衡。 营养物供应的变化导致细胞适应恢复平衡。 营养过多会导致线粒体网络断裂,从而导致线粒体的生物能效率降低。 这样可以避免能量损失。 相反,由于代谢饥饿,线粒体变长以增加其生物能效率。这些运动的诀窍是什么? 如果细胞处于饥饿状态,那么线粒体的合并可以提高其生物能效率(每个营养分子产生的ATP量)。 如果过多的营养进入细胞,则可以1)储存它们,或2)以热的形式耗散这种能量。 在这种情况下,线粒体的任务是以热的形式散发更多的能量,以ATP的形式散发更少的能量(NADH和ROS的积累会导致氧化应激)。 线粒体的碎裂使它们降低了生物能效率,其降低的主要机理被认为是质子的“泄漏”。
因此,我们开始工作,线粒体的生活在分裂和融合的循环中不断发展(图3)。
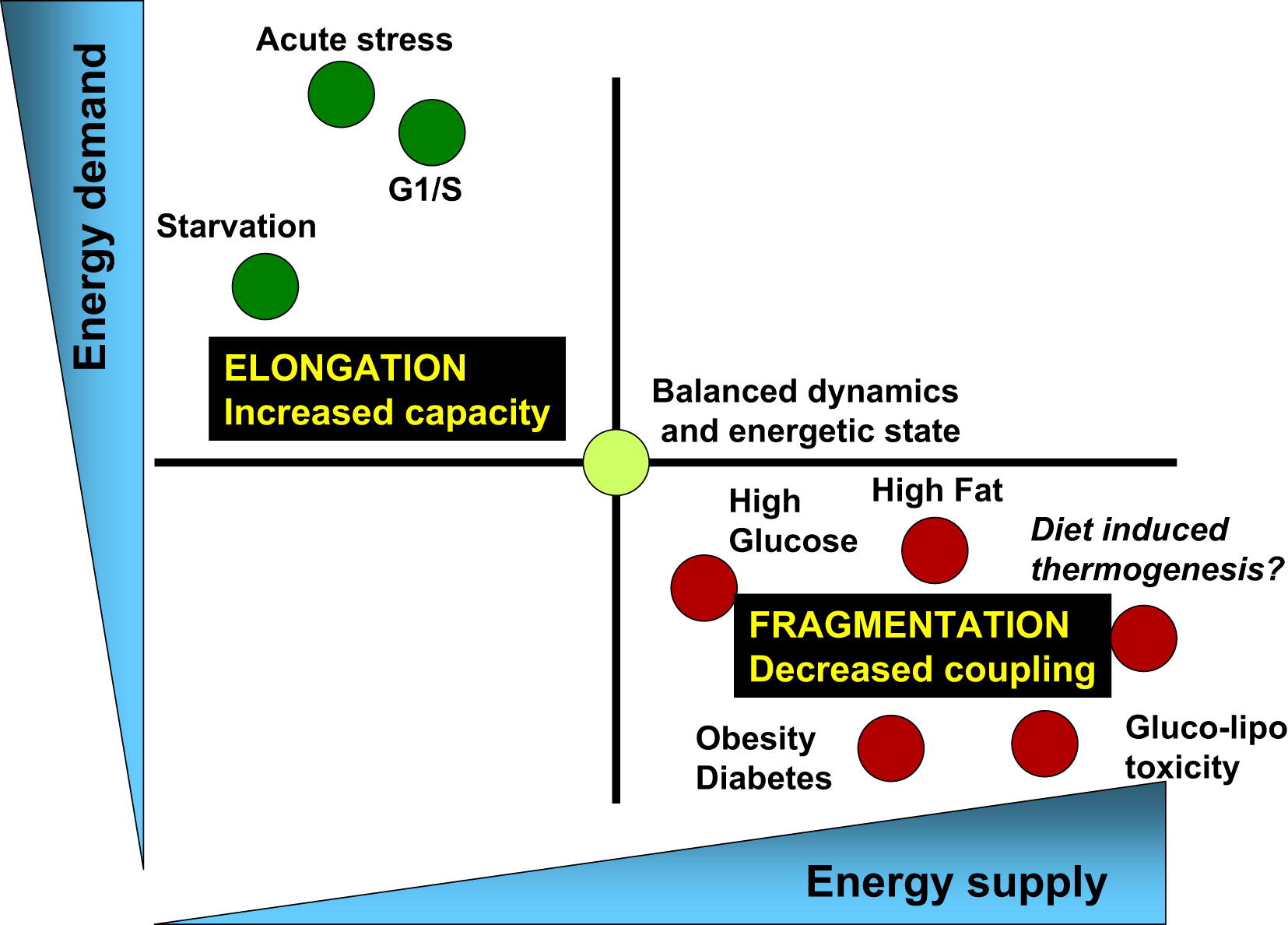 图3 能量消耗和能量供应 的 平衡与线粒体结构及其生物能效率的相应变化相关[3]
图3 能量消耗和能量供应 的 平衡与线粒体结构及其生物能效率的相应变化相关[3]
与能量需求增加和能量供应减少有关的生理过程(例如,急性应激,饥饿和G1 / S期)的特征在于线粒体延长和与ATP合成相关的呼吸作用。 另一方面,与能量需求减少和其供应增加(高水平的营养,肥胖和2型糖尿病)相关的生理过程与线粒体分裂,产热或线粒体功能降低有关。健康的裂变和融合周期是细胞代谢健康的关键
线粒体分裂和融合的正常循环是其质量控制的关键因素。 怎么了 线粒体分裂过程中,形成了两个子链,其中一个具有较高的膜电势并进一步进入融合分裂循环,而另一个具有更多去极化的膜保持分离状态,直到恢复膜电势为止。 如果电位得以恢复,它将与线粒体网络重新结合。 如果保持去极化,则在自噬过程中将其消除,这是线粒体池质量的关键(图4)。
长期抑制线粒体分裂(延长细胞饥饿)会导致线粒体受损,无法分离[3,4]。
另一方面,过量的营养物导致线粒体融合的抑制,这导致线粒体动力学循环的破坏,增加了细胞内线粒体的异质性。 是的,在食物过多的情况下,线粒体的碎裂是有保护作用的,但是延长的碎裂(如长时间的融合)对线粒体的质量控制有害。 没有选择性清除;线粒体的质量会减少,由小的去极化线粒体组成。
 图4 线粒体的生命周期及其对养分利用率的调节[3]
图4 线粒体的生命周期及其对养分利用率的调节[3]线粒体蛋白酶不仅是蛋白质
在分子水平上,线粒体融合是一个两步过程,需要在单独的顺序事件中协调外层和内层膜的融合。 在哺乳动物中,该过程由属于GTPases的三种蛋白质调节:Mfn1和Mfn2对于融合外膜是必需的,而OPA1-对于融合内膜是必需的。 分裂需要其他蛋白质,Fis1和Drp1。
在丧失功能和获得功能的研究中已经研究了线粒体蛋白的作用。 线粒体蛋白蛋白质的小鼠突变体早在妊娠中期就死亡,因为线粒体融合对它们而言变得不可能。 线粒体融合蛋白对于自噬和线粒体吞噬过程很重要。 Mfn2在心肌细胞中表达的降低阻止了自噬过程的开始,因为自噬体与溶酶体的融合被阻止了。 Mfn2的消耗导致线粒体膜电位的降低;作为补偿,呼吸链的功下降,葡萄糖摄取增加,糖原合成减少。 细胞切换为厌氧性浮细胞,这就是细胞发生肿瘤变性的途径。 Mfn2缺乏会导致神经退行性改变。 Mfn2在骨骼肌中表达的增加会增加其对胰岛素的敏感性。
Mfn1执行类似的功能,但可能在其他组织中(Mfn2和Mfn1的表达在不同的组织中有所不同)-Mfn1在心脏,肝脏,胰腺,睾丸和心脏,骨骼肌,脑,棕色脂肪组织中的表达更多。 。
因此,丝裂霉素是线粒体动力学的关键调节剂。 mitofusin的表达在不同器官中是不同的,它们提供生物能效率和对营养素可用性的适应机制,而细胞的“命运”取决于它们。 毫不奇怪,线粒体融合蛋白是药理学干预的潜在靶标[2,5]。
下丘脑,线粒体,代谢功能障碍和衰老
线粒体的动力学在所有细胞中都很重要。 在胰腺的β细胞中,线粒体是营养的传感器和胰岛素合成信号的产生者,在肌肉中,线粒体动力学对于调节葡萄糖代谢等非常重要。 但是,一个人不仅是不同类型的单元格的集合,每个单元格都做出独立的决定。 生物是具有维持能量和葡萄糖稳态的主要调节环节的系统。 这个主要的调节器是下丘脑。
下丘脑位于中脑,正是它提供了神经和体液调节系统的相互联系。 下丘脑神经元感知,处理并响应来自脂肪组织(瘦素),胰腺(胰岛素)和其他激素刺激(生长素释放肽,胆囊收缩素,胰腺多肽等)的信号。 下丘脑控制人类内分泌系统的活动,因为它的神经元能够分泌神经内分泌递质,从而刺激或抑制垂体产生激素。 换句话说,下丘脑的质量不超过大脑的5%,是调节内分泌功能和维持整个生物体内稳态的中心。
甚至Dilman(Dilman V. M“大生物钟”)也指出了下丘脑在代谢功能障碍系统性发展中的主导作用,导致肥胖,糖尿病,心血管,肿瘤疾病和衰老。 根据迪尔曼(Dilman)形成的高度适应性疾病理论,下丘脑受体对来自人体组织(瘦素,胰岛素等)的信号的敏感性会随着年龄的增长而逐渐降低。 为了激发他的“反应”,需要越来越多的一种或另一种激素-更多的胰岛素,更多的瘦素。 产生胰岛素和瘦素抵抗力,导致衰老和死亡的代谢疾病。
根据执行的功能,将神经元组合并到下丘脑的核中。 其中之一-弧形(弓形)核心是饮食行为和新陈代谢的关键调节剂。 可以在其中形成分别对应于AgRP和POMC神经元的产氧神经肽(刺激食欲)和产食神经肽(抑制食欲)。 周围信号(胰岛素,生长素释放肽,瘦蛋白等)影响刺激或抑制食欲的肽的表达,从而确保中央调节的连贯性(图5)。
 图 5. 下丘脑控制能量代谢。 大脑会整合来自胰腺,脂肪组织和胃等周围组织的代谢信号(瘦素,胰岛素,生长素释放肽,PYY3-36)。 在大脑中,专门的神经网络协调食物吸收和消耗中的适应性变化[5分]。
图 5. 下丘脑控制能量代谢。 大脑会整合来自胰腺,脂肪组织和胃等周围组织的代谢信号(瘦素,胰岛素,生长素释放肽,PYY3-36)。 在大脑中,专门的神经网络协调食物吸收和消耗中的适应性变化[5分]。那么谁以及如何调节下丘脑神经元的敏感性呢?
对脑组织中线粒体动力学的研究表明,线粒体动力学在下丘脑神经元控制体内葡萄糖水平和能量动态平衡的能力中起着重要作用[6,7,8]。
在刺激食欲并调节体重增加的AgRP神经元(促进饥饿的AgRP神经元)中,饥饿导致线粒体分裂,高脂喂养导致融合。 也就是说,线粒体的反应不同于大多数其他细胞。
MX在这些神经元中的融合调节高脂饮食对电活动的反应,从而刺激产卵肽(AgRP肽)的产生,这对于体重增加和脂肪过多的脂肪沉积是必需的。 这些神经元中Mfn1和Mfn2的缺失导致大鼠瘦素的增加,原因是循环中的瘦素水平降低。
POMC神经元(抑制食欲)具有相反的功能,线粒体对营养摄入的响应动力学也不同。 这些神经元中线粒体蛋白表达的减少导致线粒体与EPS的连接中断,结果是食欲亢进,瘦素抵抗和肥胖症。 同时,粮食消费增加,能源消耗减少。
因此,人体对高脂饮食的反应取决于下丘脑神经元线粒体动力学的模式。 神经元中线粒体的重塑提供了它们对营养摄入的反应,刺激了神经肽的产生,这会刺激或抑制食欲,从而影响人体的新陈代谢(图6)。
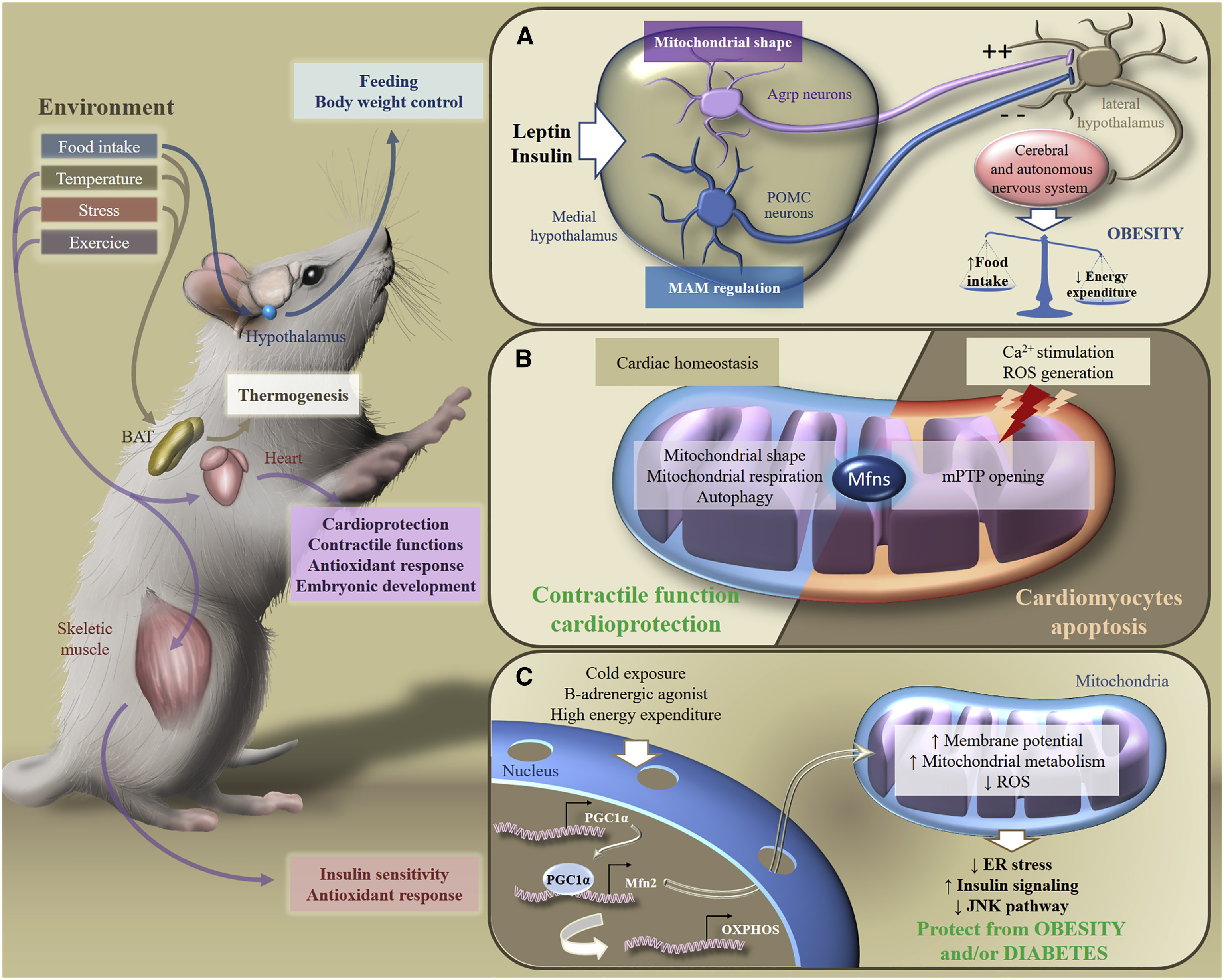 图6。 对环境刺激的代谢适应[2]
图6。 对环境刺激的代谢适应[2]响应外源性刺激,Mfns参与不同器官中代谢信号的转导,从而确保整个体内能量稳态的维持。 特别地,响应于食物摄入,温度变化,压力或运动,棕色脂肪组织,脑,心脏或骨骼肌使它们的新陈代谢适应营养,体重,收缩功能,抗氧化反应或胰岛素敏感性的控制。
如何影响线粒体的动力学?
1.营养与运动营养周期过量食物和高脂饮食(HFD)会抑制细胞中的线粒体融合(某些脑神经元的机制不同)。 不完整的线粒体分裂融合周期破坏了自噬过程→细胞内线粒体异质性增加→没有选择性去除线粒体→线粒体积聚功能障碍。
热量限制(进食/禁食周期)刺激生物能适应,提供线粒体质量机制。
2.健康的膜:硬脂酸,心磷脂,磷脂酸所有关键过程都取决于线粒体膜的“健康”状况,即自噬,线粒体吞噬,细胞凋亡,线粒体与内质网的关系以及线粒体的动力学。 细胞器的膜由脂质和蛋白质组成。 这些膜的重塑是由特定脂质和蛋白质之间的相互作用控制的。
饱和脂肪酸包括棕榈酸(C16)和硬脂酸(C18)。 结果表明,使用硬脂酸(C18:0)刺激线粒体融合过程。 其作用与对丝裂霉素的作用有关。 在小鼠中,硬脂酸的膳食补充剂可以部分恢复由Pink1或parkin基因突变引起的线粒体功能障碍。 在低C18:0饮食中连续2天的中性粒细胞中,线粒体处于碎片状态(50%的细胞破碎了MX,10%的细胞与MX连接)。 3小时后,使用硬脂酸使它们融合线粒体[8]。 因此,硬脂酸对于维持线粒体动力学很重要。 在可可豆中发现大多数硬脂酸(31-34%)。
磷脂是细胞器膜的主要成分。 它们还调节线粒体的动力学,其作用是不同的[9]。
心磷脂(CL)刺激线粒体分裂和内膜融合。
心磷脂对于电子传输链的复合物IV(柠檬酸C氧化酶)的操作是必需的。 心磷脂几乎完全位于线粒体内膜中。 随着年龄的增长,心磷脂的量会减少。 有一种理论认为,心磷脂功能的丧失与分子中的饱和脂肪酸被多不饱和脂肪酸替代有关。 为了解决这个问题,有必要将富含硬脂脂肪酸的饱和脂肪首先引入饮食中。
为了增加饱和脂肪酸向膜的递送效率,可以使用转运蛋白。 例如,使用饱和的磷脂酰胆碱(二棕榈酰磷脂酰胆碱和dyseroyl磷脂酰胆碱),有可能将饱和的FAs直接递送至心磷脂[10]。 胆碱作为载体,很容易穿过细胞质并进入线粒体。
磷脂酸(RA)抑制线粒体分裂并刺激外膜融合(图7)。
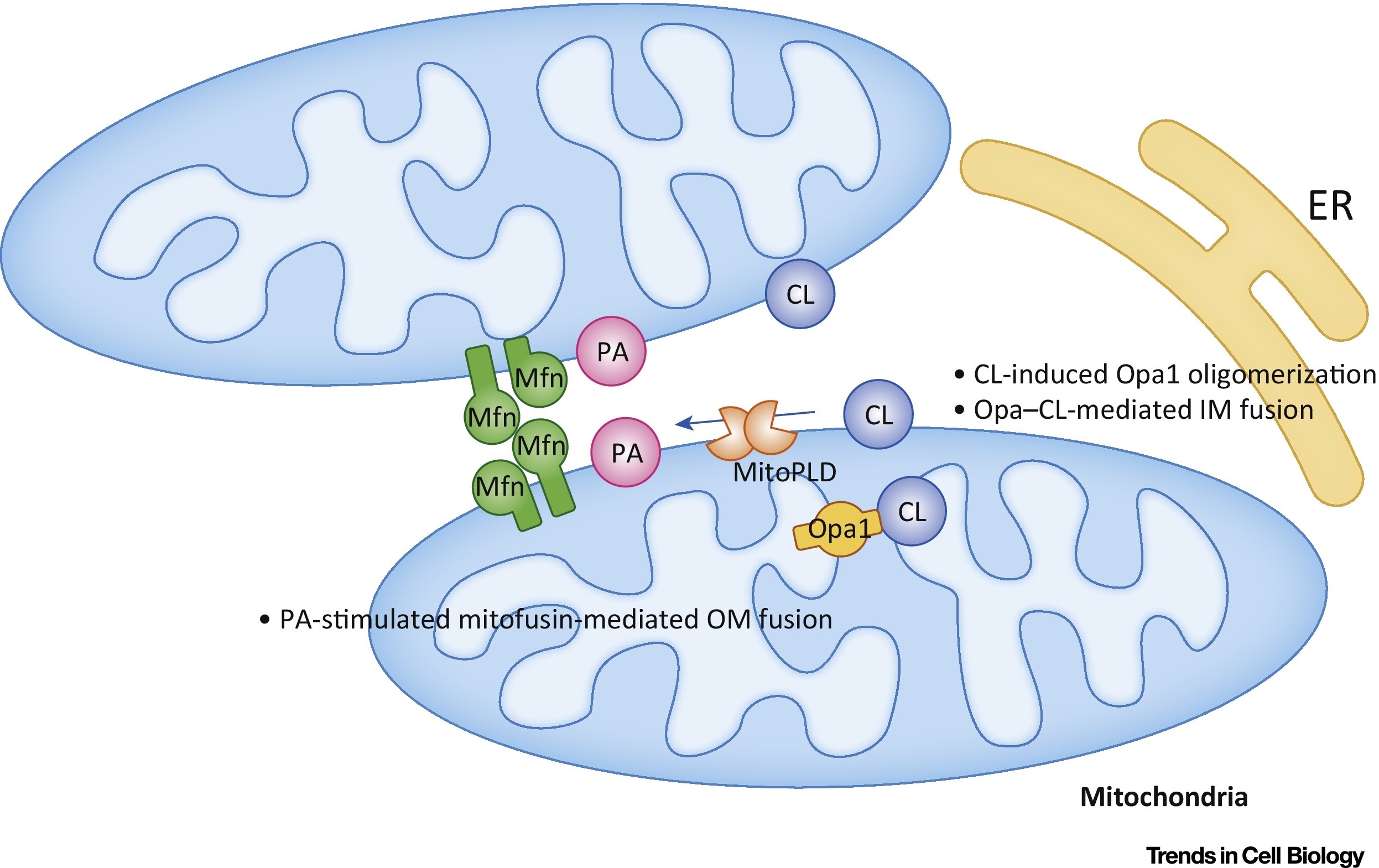 图7. 磷脂酸(PA)和心磷脂(CL)对线粒体融合的调控[9分中]。
图7. 磷脂酸(PA)和心磷脂(CL)对线粒体融合的调控[9分中]。在外膜(OM)中,RA刺激丝裂霉素介导的(Mfn)融合。 在内膜(IM)中,CL刺激Opa1介导的融合。 缩写:ER-内质网; MitoPLD-线粒体定位的磷脂酶D。
3.调节线粒体蛋白(负责线粒体动力学的蛋白质)的表达我们上面讨论的所有内容(热量限制,硬脂酸,磷脂)都通过影响丝裂霉素的表达来发挥作用。此外,还有许多药物可以间接影响线粒体的动力学。这些措施包括使用二甲双胍。最有趣的是使用会直接影响线粒体融合蛋白表达的物质。一种潜在的药物称为来氟米特(来氟米特),已被FDA批准[5,11]。它是Mfn1和Mfn2表达的诱导剂,并已注册为治疗类风湿关节炎的药物。线粒体基因疗法
线粒体动力学受损可能与负责线粒体融合和分裂的蛋白质表达受损有关。此外,这些蛋白质的功能障碍可能与它们的突变有关(而且这种情况最常见)。考虑线粒体功能障碍的因果相互作用有两种方法。以前认为,生活方式,包括暴饮暴食,会导致自由基的形成,氧化应激,线粒体基因组的突变,从而导致线粒体功能障碍。但是,最近有令人信服的证据表明,线粒体DNA的突变是不可避免的,均具有异质性DNA点突变,并且与复制错误相关,而与氧化损伤无关,而线粒体DNA相当稳定[12]。在受精卵的阶段,我们的一些线粒体带有突变。随着时间的流逝,它们分裂,有更多的突变线粒体,它们无法正常执行其功能。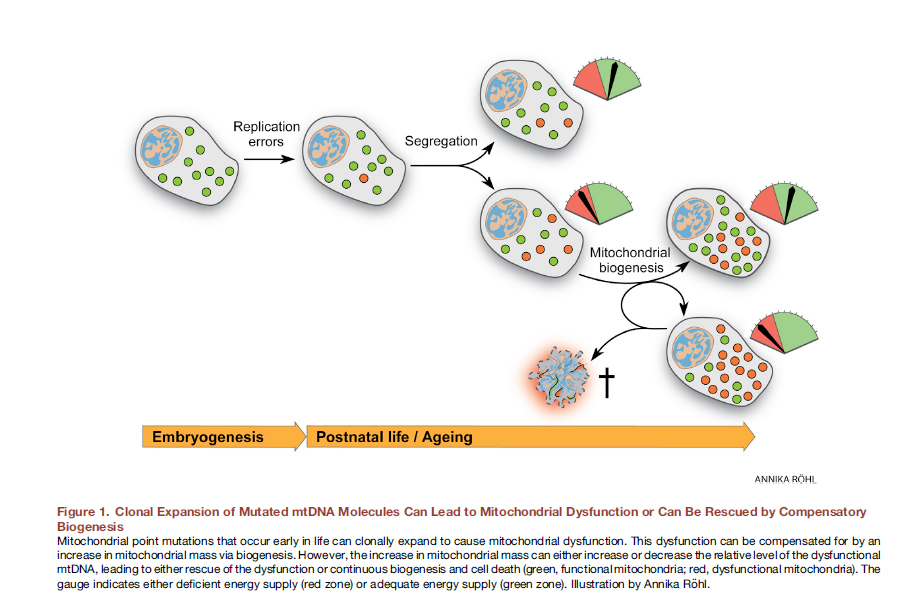 图 8 突变的mtDNA分子 的克隆扩增可导致线粒体功能障碍或可通过代偿性生物发生“保存” [12分]。在体内,线粒体基因组的编辑可能非常有用。已经显示,对于小鼠中的异质性DNA点突变,使用腺病毒载体进行靶向锌指核酸酶(mtZFN)的递送已经获得了巨大的成功[13]。
图 8 突变的mtDNA分子 的克隆扩增可导致线粒体功能障碍或可通过代偿性生物发生“保存” [12分]。在体内,线粒体基因组的编辑可能非常有用。已经显示,对于小鼠中的异质性DNA点突变,使用腺病毒载体进行靶向锌指核酸酶(mtZFN)的递送已经获得了巨大的成功[13]。线粒体转移
消除线粒体功能障碍的另一种有前途的方法是线粒体移植。这种方法的本质是用健康的外源性线粒体“替代”受损的线粒体。这种方法首先在临床上用于患有心肌缺血的儿童。使用自体分离的线粒体进行移植,将其与腹直肌分离(进行活检,然后制备制剂),然后通过直接注射进行给药[14]。引入线粒体的各种方法正在开发中:当线粒体本身“搜索”到哪个细胞时,直接注射分离的线粒体(局部注射)和全身注射到血流中。几组研究人员正在研究线粒体移植在帕金森氏病,肝缺血,中风,线粒体疾病中的可能性[15]。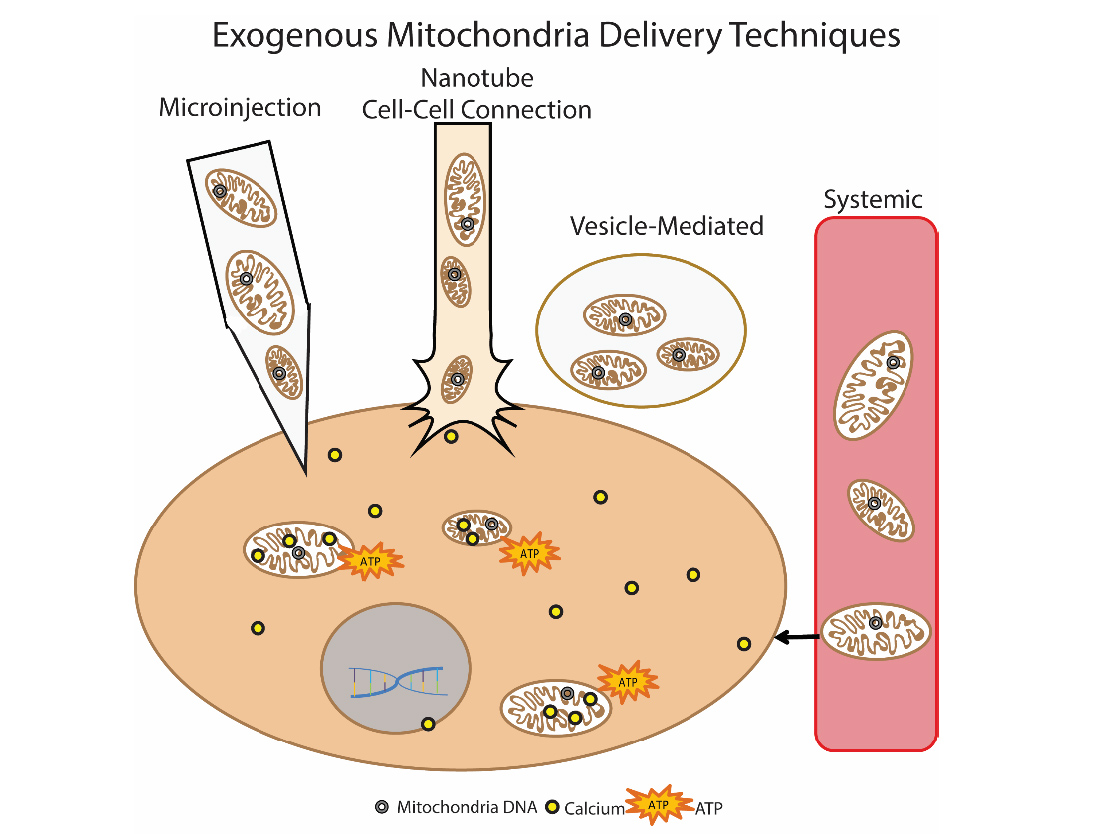 图9 外源线粒体向细胞的递送方法作者Olga Borisova
图9 外源线粒体向细胞的递送方法作者Olga Borisova文学作品1. Kauppila, Timo ES, Johanna HK Kauppila, and Nils-Göran Larsson. «Mammalian mitochondria and aging: an update.» Cell metabolism 25.1 (2017): 57-71.
www.sciencedirect.com/science/article/pii/S15504131163050222. Schrepfer, Emilie, and Luca Scorrano. «Mitofusins, from mitochondria to metabolism.» Molecular cell 61.5 (2016): 683-694.
www.sciencedirect.com/science/article/pii/S1097276516001337#fig13. Marc Liesa, Orian Shirihai “Mitochondrial Dynamics in the Regulation of Nutrient Utilization and Energy Expenditure” Cell methabolism (2013): 491-506
www.sciencedirect.com/science/article/pii/S1550413113001046#fig34. Ramos, Eduardo Silva, Nils-Göran Larsson, and Arnaud Mourier. «Bioenergetic roles of mitochondrial fusion.» Biochimica et Biophysica Acta (BBA)-Bioenergetics 1857.8 (2016): 1277-1283.
www.sciencedirect.com/science/article/pii/S00052728163008585. Cunarro, Juan, et al. «Hypothalamic mitochondrial dysfunction as a target in obesity and metabolic disease.» Frontiers in endocrinology 9 (2018): 283.
www.frontiersin.org/articles/10.3389/fendo.2018.00283/full6. Marcelo O.Dietrich et al. «Mitochondrial Dynamics Controlled by Mitofusins Regulate Agrp Neuronal Activity and Diet-Induced Obesity”.
www.sciencedirect.com/science/article/pii/S0092867413010957#figs27. Steculorum, Sophie M., and Jens C. Brüning. „Sweet mitochondrial dynamics in VMH neurons.“ Cell metabolism 23.4 (2016): 577-579.
www.sciencedirect.com/science/article/pii/S15504131163011768. Senyilmaz-Tiebe, Deniz, et al. „Dietary stearic acid regulates mitochondria in vivo in humans.“ Nature communications 9.1 (2018): 3129.
www.nature.com/articles/s41467-018-05614-69. Kameoka, Shoichiro, et al. „Phosphatidic Acid and Cardiolipin Coordinate Mitochondrial Dynamics.“ Trends in cell biology (2017).
www.sciencedirect.com/science/article/pii/S096289241730158710.
raypeatforum.com/community/threads/mitolipin-liquid-saturated-phosphatidylcholine-pc-mix.1039811. Miret-Casals, Laia, et al. „Identification of new activators of mitochondrial fusion reveals a link between mitochondrial morphology and pyrimidine metabolism.“ Cell chemical biology25.3 (2018): 268-278.
12. Kauppila, Timo ES, Johanna HK Kauppila, and Nils-Göran Larsson. „Mammalian mitochondria and aging: an update.“ Cell metabolism 25.1 (2017): 57-71.
13. Gammage et al. “Genome editing in mitochondria corrects a pathogenic mtDNA mutation in vivo” Nature medicine, 2017
www.nature.com/articles/s41591-018-0165-914. Emani, Sitaram M., et al. „Autologous mitochondrial transplantation for dysfunction after ischemia-reperfusion injury.“ The Journal of thoracic and cardiovascular surgery 154.1 (2017): 286-289.
www.jtcvs.org/article/S0022-5223 (17)30258-1/fulltext
15. McCully, James D., et al. „Mitochondrial transplantation: From animal models to clinical use in humans.“ Mitochondrion 34 (2017): 127-134.
www.sciencedirect.com/science/article/pii/S1567724917300053