引言
这句话是关于什么的
如果一个人听到“现实模拟”,那么他很可能会提出各种科幻作品(例如《黑客帝国》,《黑暗之城》或《零定理》)或电脑游戏。 如果人的头因工程技术水平而阻塞,则可能会弹出
KOMPAS-3D AutoCAD,Solid Edge或NX等软件包。 一个正在听科学的人可能会想起
各种空间装置的任何
模型 。
但是,还有一个更高的现实水平,事实证明,这一水平应该被遗忘:所有化学反应发生的那个水平是原子和分子的水平。 也可以在计算机上相当成功地进行模拟。 由于量子力学负责“现实”这一部分的所有工作,因此这种计算通常称为量子化学。 我们将讨论通过实验方法研究的现实与现实的联系。
本文将介绍最基本的内容。 但是,阅读科学期刊和收听各种报告的实践表明,应该不断提醒这一点。
本文是为那些了解和/或对原子和分子的生活感兴趣的人设计的。取自xkcd.com简要背景碰巧的是,一个不幸地在俄罗斯科学领域工作的人邀请我在他一所著名的俄罗斯物理大学的2人课程中作演讲。 但是,由于一个奇怪的巧合,她被转移到同时举行的一个学生会议上...在那里,她并没有引起学生的兴趣,我对此材料感到非常非常抱歉,因此我决定对Habr进行垃圾邮件处理,试图将这门教育讲座变成一本通俗的科学文章。
研究分子寿命的物理方法
从学校化学和物理课程中我们知道,所有物质都是由原子,分子,离子或其组合组成的。 而且我们似乎甚至不知道他们过着什么样的生活。 但是,这些信息应该有自己可靠的来源(研究方法),而且确实如此。
监视原子生命的方法有很多。 例如,那些愿意的人可以在古典教科书中更详细地了解其中的一些知识。
-彭丁·Y·A。(Vilkov L.V.) 化学物理研究方法。 -M.:Mir,2006年,
-DragoR。化学物理方法。 -M.:Mir,1981年。
但是,大致且相当容易地,3种主要方法脱颖而出:
- 光谱法
- 衍射法
- 各种显微镜检查方法(无论是半透明还是扫描,对我们来说现在都不再是必需的)。
不会讨论后者,但是它的工具并不比前两个重要。
为什么不会谈论显微镜(在显微镜下,我只是不回避这个词)
物质研究的光谱学方法
这组强大的方法为我们提供了非常非常多的东西:从在星际介质和其他行星中搜索和确定分子,到在机场进行常规爆炸物检查。
光谱方法的一般原理
在谈论光谱学时,通常指的是以下一般操作原理。
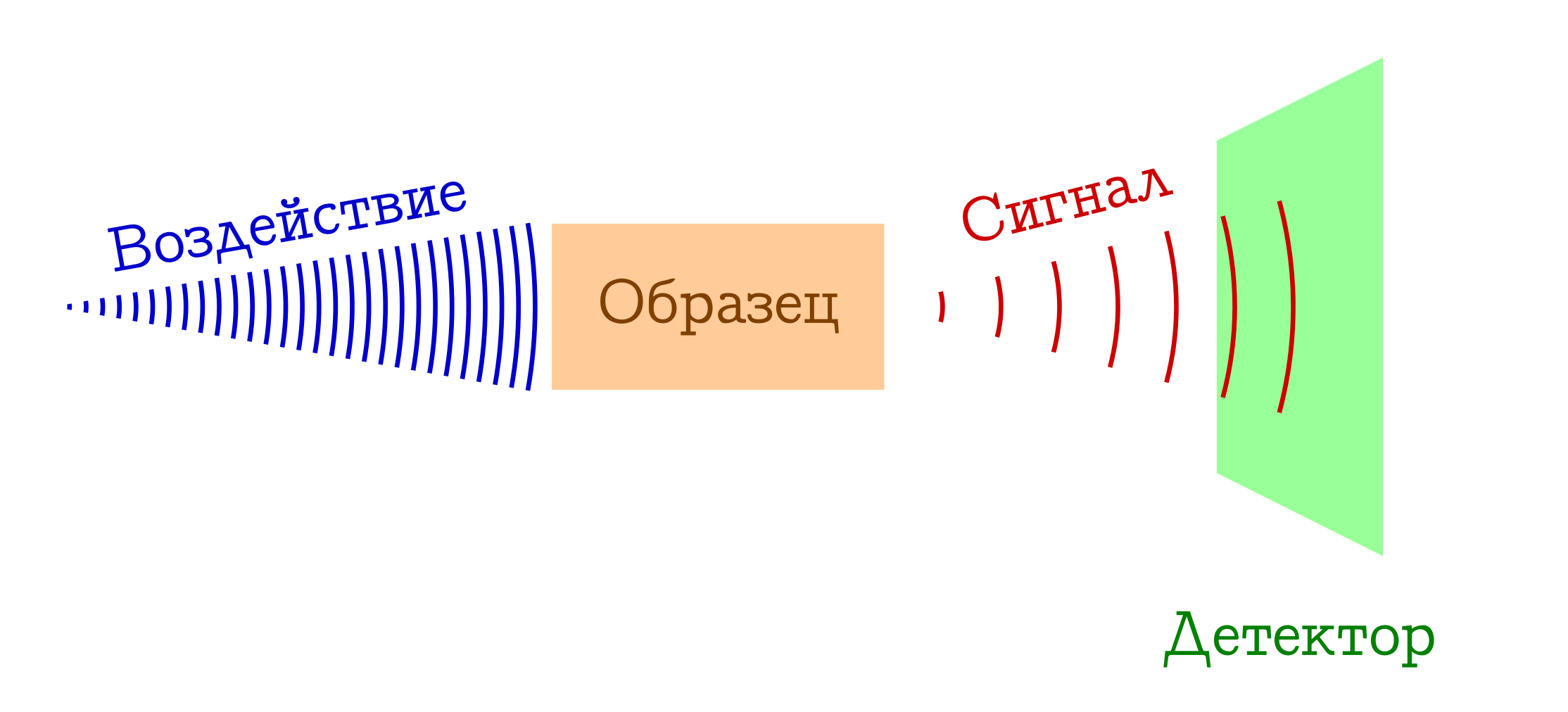
研究物质的光谱方法的一般方案
- 我们有一些东西(例如灯泡/激光/太阳光)作用于我们感兴趣的样本。 大多数情况下,这是电磁研究,但很可能是电子(例如,在通过电子撞击发生电离的质谱中)或血浆中可能存在和不可能的混合物(例如,在火焰光谱中 ,受到学童和化学学院的初中生的喜爱)。 一种或另一种方式,必须对我们的样本起作用。
- 当暴露于样品时,会发生某些改变其状态的事情。 这可能是过渡到某种激发水平(在任何分光光度法或拉曼光谱法中),甚至是分子系统的崩溃(如在质谱 法或光电子光谱法中 )。 但是在某种程度上,模式应该有所不同。
- ???
- 利润!!! 我们在分子水平上随样品的这种变化记录某种信号(发射或吸收)。 这可能会丢失用于更换样品的光子(然后我们有了吸收光谱),反之亦然,可能是物质初步激发后发射的过量光子(发射光谱),由于与物质相互作用而导致初始光子波长的变化(拉曼光谱,更多)。在国外被称为
Ramenovskaya Ramanova ),或原始分子的愚蠢片段(如质谱或光电子能谱 )。 有很多选择-本质是相同的:有信号!
例如,您可以引用许多不同的字母:NMR,ESR,MW,THz,IR,UV / Vis,XRF,MS,PES,EXAFS,XANES等。 等
所有(或其中许多)对每个化学家都是熟悉的(或应该熟悉的)。 所有这些方法都是自重研究物质的研究人员的标准武器库(绝不完全)。
光谱范围及其与分子寿命的关系
 取自xkcd.com
取自xkcd.com由于在绝大多数情况下,光谱学仍与电磁辐射有关,因此将电磁波谱的范围与原子分子寿命的各个方面联系起来是合乎逻辑的。 毕竟,光谱学中使用的电磁波频率是一种“时钟”,可让您检测分子系统中该过程持续的时间。 因此,更改此频率,您可以研究(甚至采取行动)不同的分子过程。
这样啊
- 从化学角度来看,在超长波长范围内没有发生任何有趣的事情,因此您无法记住它。
- 随着无线电和微波频率的变化(甚至是长波红外辐射,IR = IR),不同的分子在气相中旋转:大而沉重-在无线电波范围内(较低的频率),小而轻-在IR中(较高的频率)。
- 但是,在IR中,(主要)会发生各种分子振动:分子内部的所有构象运动和其他非显而易见的运动都在长波长IR中发生,而拉伸振动(拉伸-化学键长度的缩短)则在短波长中发生(最大4000 cm –1 )。
- 好吧,接下来是光谱的地方,那里有各种电子跃迁存在(直到γ量子区)。 在较低的频率下(可见光,UV =紫外线和柔和的X射线),与价电子相关的跃迁主要存在。
我们为什么看到?顺便说一句,正是由于我们可以看到电子跃迁:在我们的眼睛(视锥细胞)中,存在着具有
视网膜成分的结构。 当可见光子被该分子吸收时,其中的双键断裂,导致顺反异构化。 正是这种变化被我们视为主要信号,然后传递到我们的大脑。
但是随着光子能量的增加(即频率增加),正如我们从普朗克公式中得出的那样 E = h n u ),我们会深入电子结构的各个层,直到我们在X射线范围内停留到最终的1s壳( 或称为X射线,K )为止。
因此,选择合适的电磁辐射波长,我们可以更详细地研究分子中的特定过程。
物质衍射法
现在让我们谈谈衍射。 这种实验的示意图也很简单。
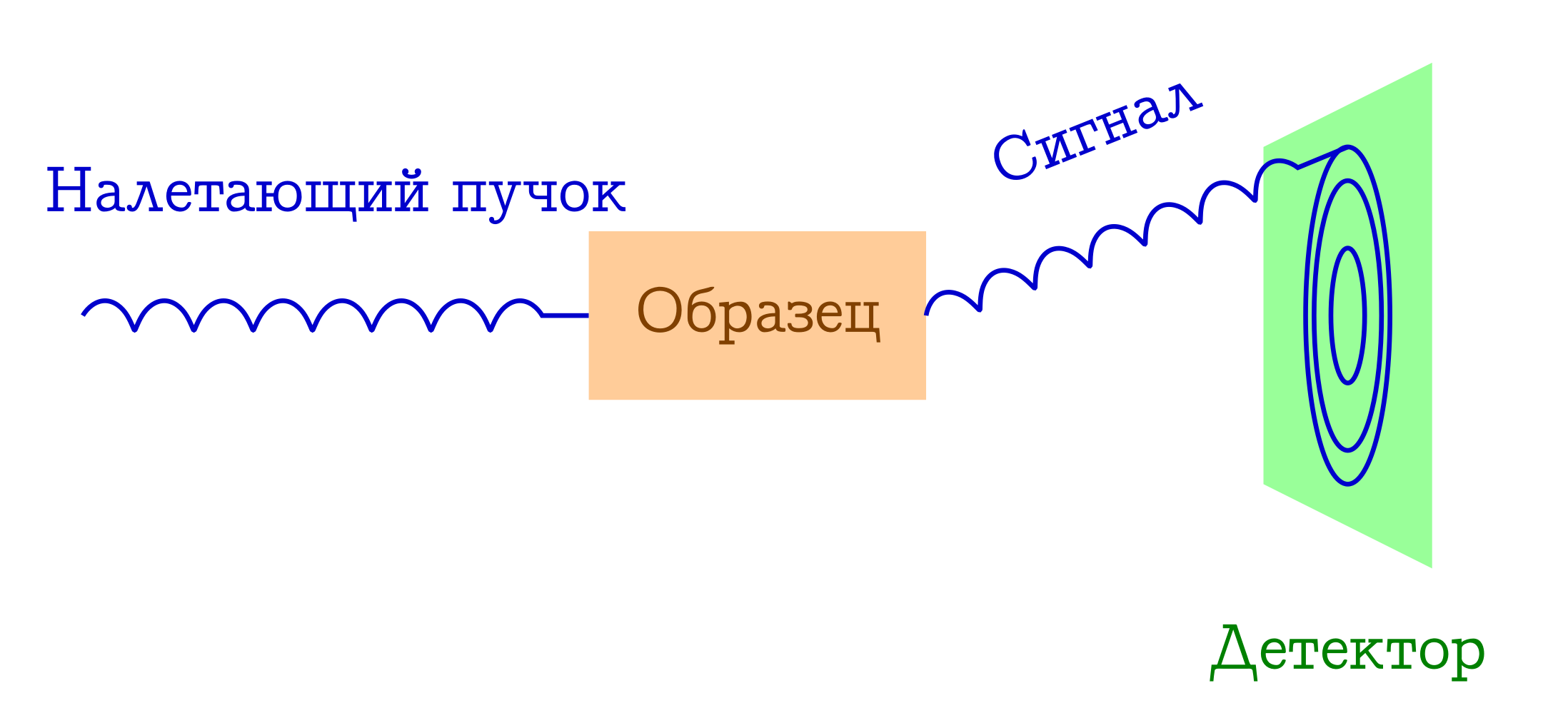
研究物质的衍射方法的一般方案
- 一束一些粒子飞到样品上。 最常见的是X射线光子,电子或中子。
- 这些粒子通过不同的机制弹性散射到我们感兴趣的样本中的原子上(即,在不改变波的波长和相位的情况下,它们只是改变其飞行方向)。 这些入射粒子对样品没有任何反应:根本没有时间对其进行反应。
- 原子间距离用作入射光束的衍射光栅,因此,结果是,我们将在检测器上看到漂亮的衍射图像。
从最后一段开始,入射粒子的波长条件(λ)出现了:它应等于或小于原子间距离的特征量级,因此这些方法的典型λ为1-0.01。
比较实验和理论计算时的主要错误类型
结果,我们得到了一张非常有趣的图片:在光谱学和衍射中,我们观察到某种左信号,该信号以某种方式
间接指示了分子系统中实际发生的事情。
与柏拉图洞穴相似这幅画使人回想起
柏拉图洞穴的
神话 。 我们有一定的分子现实世界。 但是,我们只能在洞穴壁(检测器)上看到阴影,这是现实级别所有有趣事物的不完整显示。
但是,幸运的是,有时我们可以从理论上计算出我们感兴趣的信号(例如,在微波,红外或紫外/可见光谱中),有时我们可以从观察到的信号中提取可用于量子化学计算的感兴趣量(例如,分子中原子之间的距离,偶极矩等)。 在这里,我们有机会将数值实验与实际实验结合在一起,进行相互比较的热情分析……而在这里,有四种类型的错误可能作为标准发生。
注意! 这里的“错误”一词并不意味着比较的结果显然是错误的。 只是比较的基础变得非常摇摇欲坠,而且草率的一步很容易弄乱整个工作。
- 实验和/或计算的不同条件 (聚集状态,温度,压力等)。 由于某种原因,我们可能突然开始比较它们之间的不同系统,因为它们是相同的。 例如,很明显,向一杯茶中添加一或五茶匙糖将导致相同的物理系统,即“糖茶”,但该系统的性质将有很大不同。 而且它很容易测量。 例如,用温度计(在糖溶解后立即测量茶的温度)或用舌头(所谓的感官分析方法之一)。 因此,在相互比较最终系统时(无论是一杯真正的糖茶还是其计算机模型),我们都不能忘记相似之处有其局限性,并且如果我们减少“相似性”的误差范围,我们最终会发现差异。
- 参数的物理和/或数学含义不同 (通常意义上的物理含义参数甚至可能不存在)。 在这里,一切也很简单:如果我们比较两个具有相似名称的数量,这并不意味着这些数量具有相同的物理含义。 例如,该城市总人口与 仅在阿妈中评级。 无论是这个等级还是那个等级(无论是多少),这些数字(或其他数字)甚至可以彼此高度相关,但是这些参数的含义仍然不同,并且可以检测到这种差异。
- “随机”错误 。 这包括实验者/模拟器理论家不知道的一些系统错误,或实验/计算中无法控制和/或预测的真正随机错误。 原则上,这些事物本身可以成为研究各种有趣的系统效果的主题。
或仅是最有用的信噪比(“信噪比”)的估算值。 - 最后一个标准错误是实验者/计算器的手从骨盆骨骼的生长 ,即普通的人为错误。 无需进行任何检查,只需仔细检查工作或重复实验即可找到并消除相应的门框。
关于后两种错误,没有什么比这更具体的了,但是关于前两种错误,如果采用特定的研究方法,您可以说很多话。 因此,我们将专注于它们。 在这种情况下,主要重点是分子的结构差异。
错误#1。 不同条件下分子性质的差异
NaCl:无错误时
出于某种原因,没有人会说氯化钠(NaCl)的单晶是一个Na
+和Cl-离子的大分子,而在疯狂的温度下通过蒸发该晶体而获得的双原子NaCl分子具有相同的我们说一下结构。
即使我们假设氯和钠之间的距离(
r NaCl )至少在这里和那里都相同,实验仍将我们放在适当的位置:
我们在哪里犯错实际上,通过这样的比较,我们允许出现错误#2,但是在这里一切都很好,如果我们评估这样的比较的误差,它们的误差约为0.01Å,这大大小于比较参数的差异。 即 这不是错误,而是真正的效果。
如何自己获取盐晶体中钠阳离子和氯阴离子之间的距离从实验数据获得双原子NaCl分子中原子之间的距离也不是那么复杂的过程。 但是问题仅仅是这样的实验是一件复杂的事情。 因此,使用已经给出所需距离的数据库更容易。
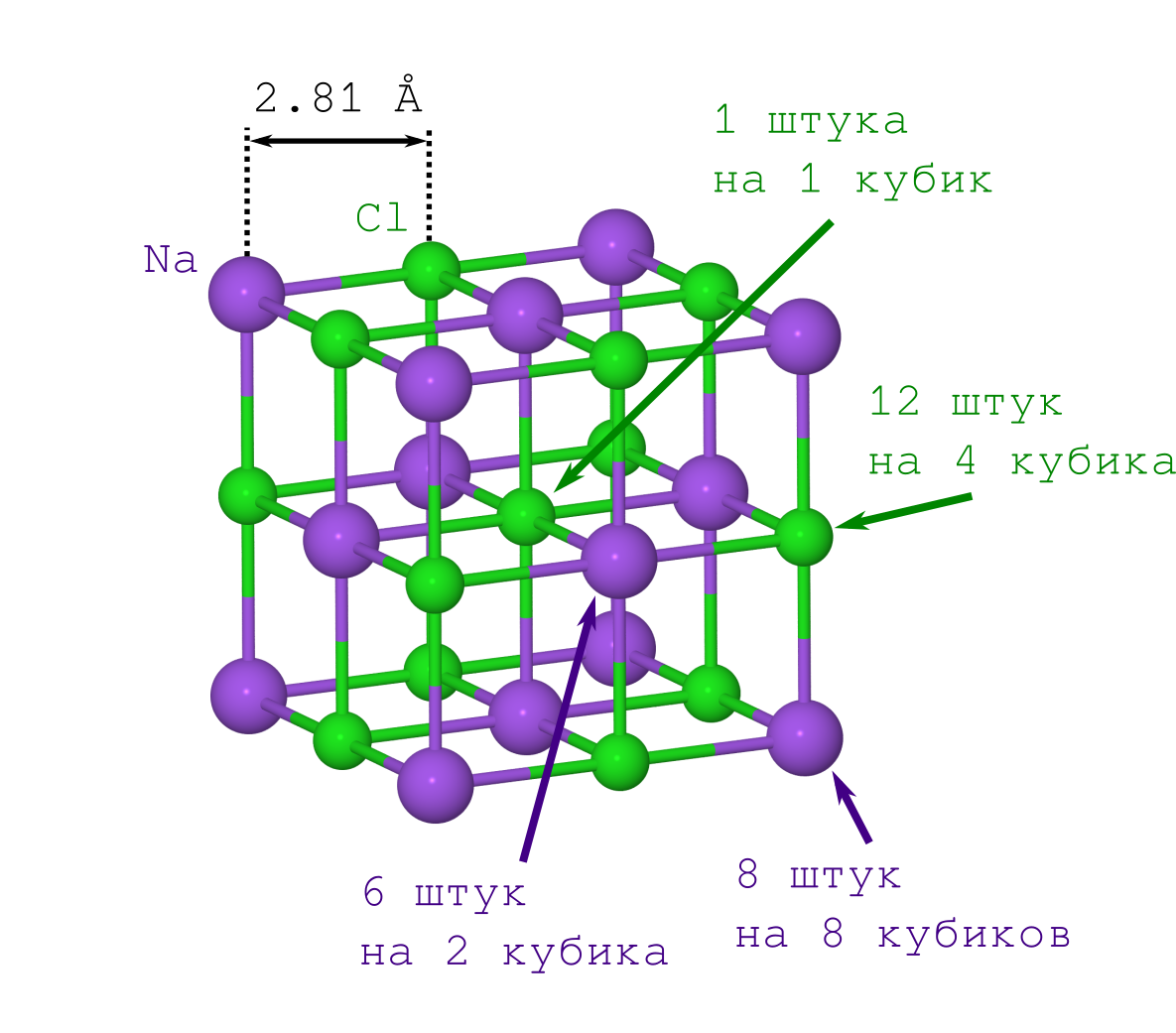
但是要获得晶体中原子之间的距离,仅表ρ= 2.165 g / cm
3的结晶盐的密度
就足够了,可以
从Wikipedia轻松获得并在家中进行测量。
要计算距离,我们需要:
如果您是第一次这样做(例如,在20世纪初),那么您将不得不承受第二点的痛苦。 但这已为现代人所知:NaCl晶格具有立方体形状,其中Na
+和Cl-离子彼此交替(请参见上图)。 通过将指示的晶体碎片相乘(“复制-粘贴”指定的片段并将其面对面调整为先前的迭代),我们可以得到任何所需大小和任何所需(矿物模型)形状的NaCl晶体。
因此,此立方体的密度应与整个晶体的密度相同。 鉴于密度为
rho= fracmV (即每体积的质量),事实证明,知道质量和体积的几何表达式,我们可以计算原子之间的距离。
立方体的体积很明显:肋骨的长度是Na-Cl(
L=2r mathrmNaCl ),表示所需的音量为
V=L3=8r mathrmNaCl3 。
质量不是那么简单。 我们大多数原子位于立方体的顶点,边缘和面上,这意味着它们同时属于这些立方体中的几个。 在计算中必须考虑到这一点。
让我们从Na
+离子开始。 我们只有两种类型(请参见晶格图案):
- 位于多维数据集的顶点的那些(与多维数据集的顶点一样多,即8个,并且它们同时位于8个多维数据集,因此您需要将此数字除以8)
- 那些躺在脸上(有6个,同时属于2个立方体)。
结果,我们得到我们的多维数据集包含
8 cdot frac18+6 cdot frac12=4 钠离子。
现在关于Cl-。 它们也只有2种类型(请参见晶格图案):
- 那些位于立方体边缘的对象(有12个,并且由4个立方体共同拥有),
- Cl-位于立方体的中心,它是1,仅属于我们的立方体。
因此,我们的多维数据集包含
12 cdot frac14+1 cdot frac11=4 氯离子。
显然,晶体的组成与NaCl的化学式相对应,但是我们的立方体的质量是相等的(不要忘记,周期表中
原子的质量是以
原子质量单位给出的):
m=4 cdot(\底行M mathrmNa23 \文字am+\底行M mathrmCl35.5 \文字amu)=234 \文本amu=234 cdot1.66 cdot10−24 textr=3.88 cdot10−22 \文字g\。
现在从关系
rho= fracmV 我们可以为长度做一个方程
r mathrmNaCl :
r mathrmNaCl3=\左( fracm8 rho\右) ,
这很容易解决:
r mathrmNaCl=\左( fracm8 rho\右)1/3=\左( frac3.88 cdot10−22 [ textg]8 cdot2.17 [\文字g/\文字cm3]\右)1/3=2.82 cdot10−8 [ textcm]=2.82 [ textÅ] 。
根据x射线晶体学2.81Å的数据(例如,来自
Abrahams,SC; Bernstein,JL的自动衍射仪的精度。氯化钠结构因子的测量// Acta Crystallographica(1965)18,926-932 ),我们仅漏掉了
0.01Å ,这很酷。
有人可能认为0.45Å的差异微不足道,但这几乎是玻尔半径(0.52Å),它等于电子最可能的距离,并且按照原子标准,该差异很大。
为什么2原子分子形式的NaCl与晶体不同这里的一切都很简单。 无限晶格为每个氯原子3s
1个钠电子产生不可逆的“跳跃”的可能性,因为由此产生的电荷差可通过与相邻原子的相互作用得到补偿。
3s
1 ( ), ,
«» :
Na:Cl↔Na+Cl−
() (), .
,
±1 , , .
NaCl (2.36 Å),
d=q⋅rNaCl=9.0 [] 在哪里
q≥0 (
+q ,
−q )
, « » 0.21, ..
d=0.21⋅9.0=1.9 [qe⋅Å] , :
q=drNaCl=1.92.36=0.8 。 «» 0.2 NaCl NaCl .
二茂铁
从离子晶体转变为分子紧密堆积的分子晶体是值得的,因此突然之间就可以毫无保留地进行比较了。但是,差异不可忘记。在这个问题上甚至有一个经典的例子:二茂铁分子。这是最简单的三明治连接。在其中,中性铁原子(像炸肉排)夹在两个五元芳香环(泡孔)之间。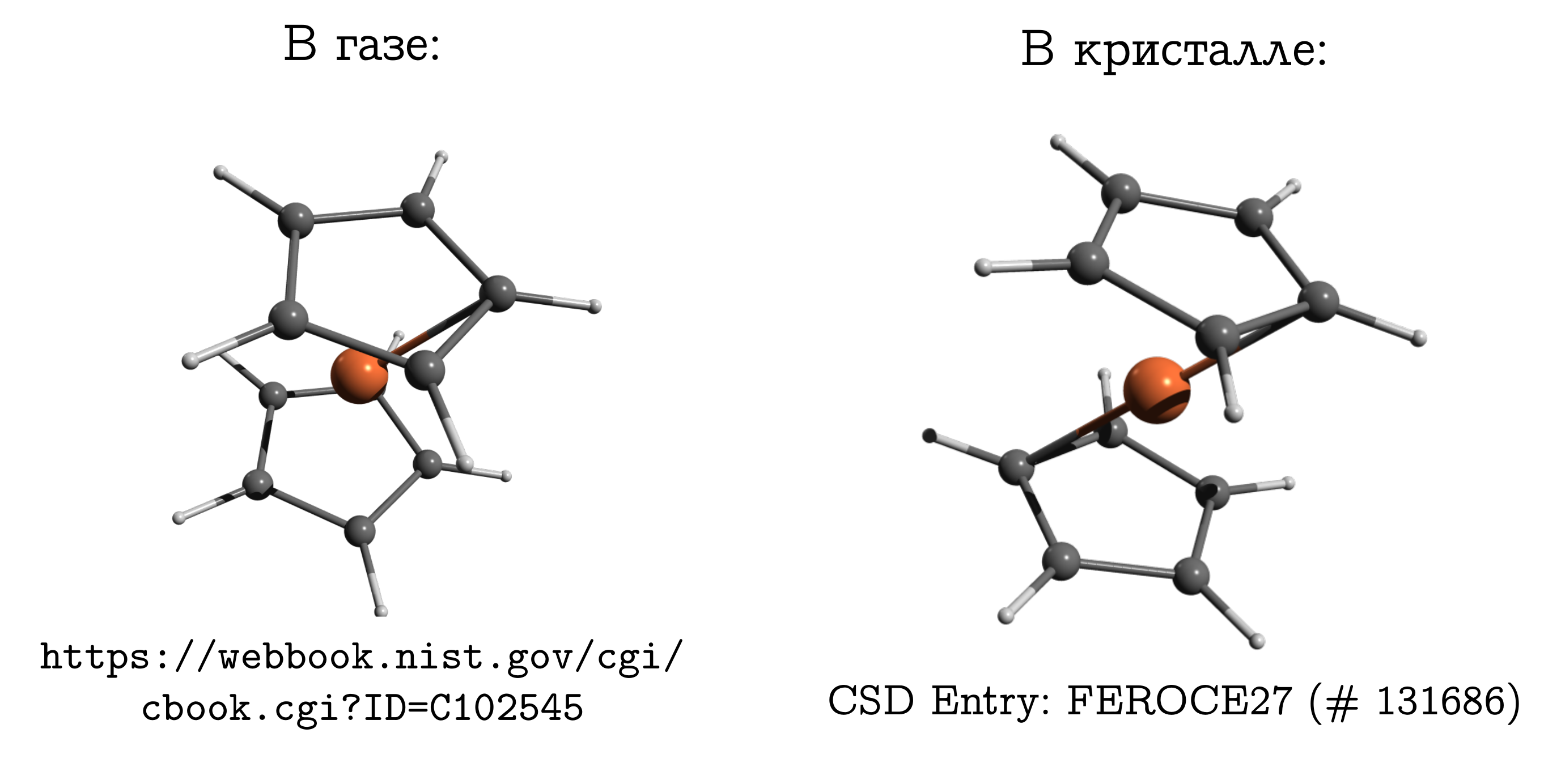 该分子可以很容易地蒸发,并发现气相中所谓的最稳定的结构是构象受阻。其中,上环和下环的碳原子和氢原子彼此相对(请参见上图),因为在这种情况下,色散相互作用最强 在分子的这些部分之间,分散总是有益的。如果我们采用二茂铁晶体,那么事实证明那里的分子具有不同的稳定构象(被烃抑制),其中一个环的氢和碳在另一个环的C-C键上方/下方。分子之间也存在分散相互作用,并且对于分子结构而言,类似的,看似不便的是由于以下事实:分子更容易仅以不舒适的形式紧密贴合在一起,并且这种个人不便可以通过相互作用来弥补。
该分子可以很容易地蒸发,并发现气相中所谓的最稳定的结构是构象受阻。其中,上环和下环的碳原子和氢原子彼此相对(请参见上图),因为在这种情况下,色散相互作用最强 在分子的这些部分之间,分散总是有益的。如果我们采用二茂铁晶体,那么事实证明那里的分子具有不同的稳定构象(被烃抑制),其中一个环的氢和碳在另一个环的C-C键上方/下方。分子之间也存在分散相互作用,并且对于分子结构而言,类似的,看似不便的是由于以下事实:分子更容易仅以不舒适的形式紧密贴合在一起,并且这种个人不便可以通过相互作用来弥补。为什么二茂铁与乙烷如此不同熟悉化学的人通常必须压倒性才能记住气体中二茂铁的结构。 毕竟,他拥有乙烷(C
2 H
6 )的记忆,其中最稳定的构象被抑制(当一个CH
3的氢位于另一个CH
3的氢“之间”) 在该位置,氢电子壳之间的原子间排斥被最小化。

改编自
www.chem.msu.su/rus/teaching/stereo而这里的全部区别在于距离。 色散相互作用势的标准形式是Lennard-Jones势(顺便说一下,这是一个,而不是两个人):
V mathrmLJ(r)= fracAr12− fracBr6
其中,第一项来自原子间排斥,第二项来自电子密度波动引起的原子间吸引力。 通常,这种潜力看起来像这样:
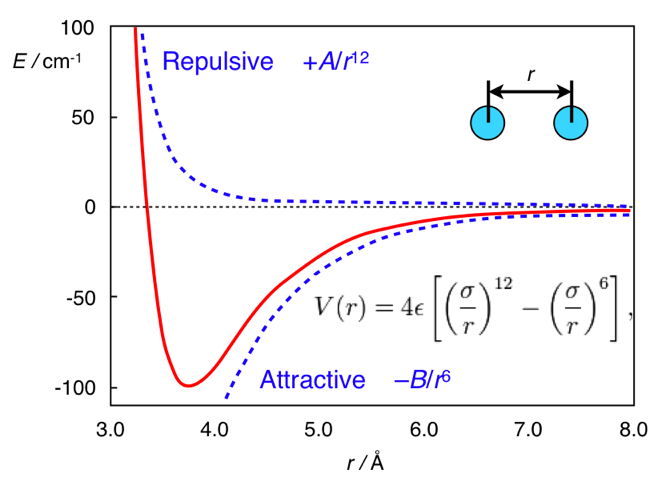
伦纳德·琼斯的潜力。 改编自
chemistry.stackexchange.com/questions/34214/双键势阱在
量子键中的物理意义在乙烷的情况下,氢原子彼此太靠近,因此它们(相对于其最小值)在曲线的左侧,并且具有排斥性。 在二茂铁的情况下,在环之间有一个相当坚固的层(铁原子),因此,环足够远,不会感到原子间的排斥。 因此,它们处于潜力的正确(有吸引力)部分。
组胺
在二茂铁的情况下,我们看到了所谓的 构象差异:分子保持不变(即没有化学键断裂或形成),并且其形状略有变化。
但是,例如,如果所谓的
互变异构转化 。 互变异构是一类化学反应,它如此容易且快速地发生,结果,我们可以同时具有一个分子的多个异构体,彼此容易相互传递。 这些异构体称为互变异构体。
一个标准的例子:酮中的酮-烯醇互变异构:

最常见的是,如本例所示,互变异构与质子从一个温暖的地方跳到另一个地方有关。 这些反应与
隧道效应有关 ,氢是最轻的原子,最容易受到该
效应的影响。
这种化学转化是许多生物分子的特征,例如
构成DNA的
含氮碱基或
糖 。
但是,当从一个系统转移到另一个系统时,此类反应的平衡常数通常会发生变化,因此在不同的阶段我们可以观察到不同的互变异构体组成。
组胺分子就是一个例子(见下图)。
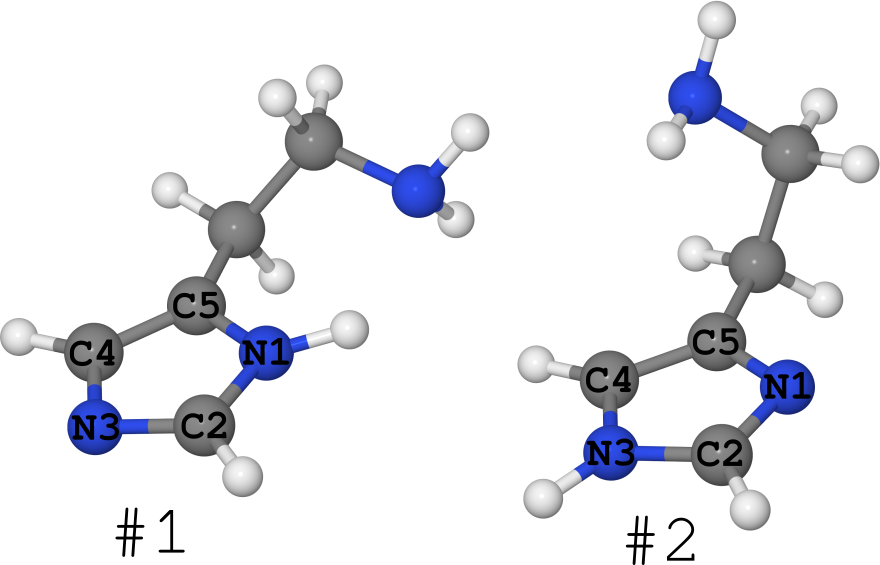
它以2个互变异构体的形式存在(我通常对构象异构体的数目保持沉默,其中有很多):
碰巧的是,对于该分子,其不同相的结构是已知的。
即 不同的相包含不同数量的不同分子,这意味着它们是不同的系统。
错误结论1

从以上示例中得出的主要结论如下:
在将一个阶段的计算与另一阶段的实验进行比较时,必须准备好系统的差异。
这并不意味着没有必要进行比较:有必要进行比较,但是只是有必要对发现的差异和/或巧合有更多的批评,并在可能的情况下评估这种影响。
错误#2。 “ Zoo”分子参数。
第二个错误的简要描述如下:如果参数的调用方式相似但不相同,则它们是不同的参数。
为了理解理论与实验之间的分歧的根源,人们将必须更详细地分析用于获得分子参数的标准实验方法和仅从理论上计算相似量的模型。
在这里,我们将再次仅讨论结构。
如何获得实验性分子结构
为了以某种方式限制自己,我们将仅讨论研究单分子结构的方法,即 关于气相。
我们有两种主要的此类信息来源:
我们将更详细地介绍每种方法。
气体电子衍射
该方法相当古老,它起源于20世纪30年代,当时德国科学家Mark和Wirl进行了关于电子在气体中的衍射的第一个实验。
很少有人知道,但是这种研究方法涉及获得三项诺贝尔化学奖。
3名贵族带电子照相输入- 彼得·德拜(Peter Debye)于1936年获得以下奖项:
“ [通过他对偶极矩的研究以及气体 中 X射线和电子的衍射研究分子结构]
这是唯一获得气体电子衍射技术的获奖者,并非没有理由。 分子散射强度的基本电子衍射方程式称为Debye。
实际上,德拜方程Iij(s)=gij frac sin(srij)rij
在这里
Iij 表示电子(或X射线或其他粒子)被第
i个和第
j个原子对
隔开一定距离的散射强度
rij 分开
s= frac2 pi lambda sin left( frac theta2 right) 散射坐标与散射角相关吗
theta 和粒子波长
lambda 和
g -这对原子散射衍射粒子的能力。
尽管事实上人们对这一奇妙的物理学( 离子溶液 的模型,用于计算晶体的热容量的模型 )没有任何印象 ,但没有记住电子衍射,但他因此而获得了主要的科学奖。
- 1954年的莱纳斯·鲍林(Linus Pauling)。是的,他曾两次获得诺贝尔奖,
甚至将维生素C (大鲍林)推向了全世界 。 在Kaltech期间,他特别参与了气体电子衍射(例如,参见DOI:10.1021 / ja01873a047 )。 当然,对自由分子的结构化学的了解帮助他创建了著名的化学键合理论 (但在这里,不要让他的晶体学背景淡化)。 - 奇怪的哈塞尔,1969年获奖者。 他因发现构象平衡而获得1/2诺贝尔奖。 他是根据对环己烷的电子衍射研究完成的。 该分子以两种构象的形式存在:椅子(椅子)和浴室(按照英国的传统,是船,船)。

从这里: www.shapeways.com/product/N5FE298DS/cyclo己烷-2-molecules-boat-and-chair-form
这些关于原子排列的选择很快就变成了另一种选择,但当时他们对此一无所知,并认为应该只实现其中一种结构。 只有电子衍射信号不想用这些结构中的任何一个来描述,只有来自这两种构象的信号组合才能解释观察到的衍射图样(有关更多信息,请参见I. Khargittai的书“ Frank Science。与著名化学家的对话”)。
该方法本身的方案非常简单(请参见下图)。
事情在真空中发生。
- 快速电子不断从阴极中击出,在阳极场中被加速到40-60 keV的能量。
- 足够散射(但很快)的电子被磁透镜聚焦,然后它们变成窄束。
- 垂直于光束安装一个装有物质的腔室。 将样品加热至沸腾,并且所产生的蒸气与电子束接触。
- 电子被分子成功散射,然后悄悄飞走,落在胶片上。
- 通常在电影前面放所谓的。 扇区设备。 这是一个旋转速度非常快的异常形状的屏幕。 事实是,电子有可能偏离其原始方向(有较大的散射角) theta ),跌落很快。 因此,为了消除强度下降,该扇形均匀地遮盖了薄膜的中央部分,而使远端部分敞开。 结果是更均匀地照亮了图片。
- 束流陷阱捕获了根本没有散射的那些电子(并且有很多)。
- 好吧,这样分子就不会在整个装置上飞来飞去,弄脏了它们,它们被冷冻在由液氮冷却的冷阱中。
结果是Debye方程描述的同心环的衍射图相同(这是一个信号)。 然后可以直接从中提取各种分子参数。
在哪里可以找到气体电子实验室?剩下的不多了。
但是在俄罗斯有两个人:莫斯科(莫斯科国立大学化学系)和伊万诺沃化学技术大学。
微波光谱
这种研究分子的方法已广为人知,因此,以最现代的修改为例,我将更简单地讨论它:傅立叶变换光谱仪(如俄语,简称为傅立叶变换微波光谱法)。
这里的设计已经更加复杂,因为它需要大量不同的电子设备(放大器,频率调制器等)。 我们将忽略所有这些,仅谈论真空室内发生的事情。
- 彼此相对,有两个喇叭天线 (就像打开遗物研究的那个一样)。 其中一个用作发送器,第二个用作接收器。
- 与这些天线垂直的是一个阀,可以发射样品。 最常见的是,它以绝热膨胀模式与某种载气(通常是惰性气体)一起以蒸汽形式发射。 在这种条件下,分子迅速冷却至接近0 K的温度,这大大简化了光谱,使其更易于解释。
- 当分子充满整个腔室时,发射天线会以线性调频信号照射它们。 在频率表示中,这对应于某个范围内所有频率的总和。
- 一些分子以不同的透射频率吸收该辐射,并进入激发态。 但是,经过一段时间后,它们会回落,开始辐射在发射天线的脉冲中捕获的东西。 这个失速看起来像是一个减小的振荡信号( 自由感应衰减 )。 第二天线也将其注册。 然后,在及时记录该信号的傅立叶变换之后,获得通常的频谱。
与电子衍射不同,电子衍射并不重要,要考虑哪种分子,在微波光谱学中,一个分子必须具有恒定的偶极矩(在极少数情况下,磁偶极矩也是合适的,这对于自由基(例如O
2分子)来说是典型的)。 这里的信号是“发射强度与 频率。” 通过一些模型从这些光谱中提取旋转常数,然后从中提取分子结构。
欢迎来到分子参数动物园!
现在是时候看看我们可以从各种实验中获得什么几何参数了。 实际上,每种数量的量都表明使用了哪种模型来拟合实验信号(通常采用最小二乘法)。 这些参数大多数都可以在Kuchitsu K.,Cyvin SJ //分子结构和振动/ Cyvin SJ(Ed。)-阿姆斯特丹:Elsevier,1972.-Ch.12中找到。 -第183-211页。
让我们重新开始电子照相。
- rg= langler rangleT 结构。 这只是给定温度下原子间距离的一组平均值。
- ra= langler−1 rangle−1T 这个值是相似的 rg ,但对于描述衍射图则更为自然。
- r alpha=rh,0=? 。 该值没有明确的物理含义,并且完全与解释模型联系在一起。 实际上,这是在晶体学中观察到的。
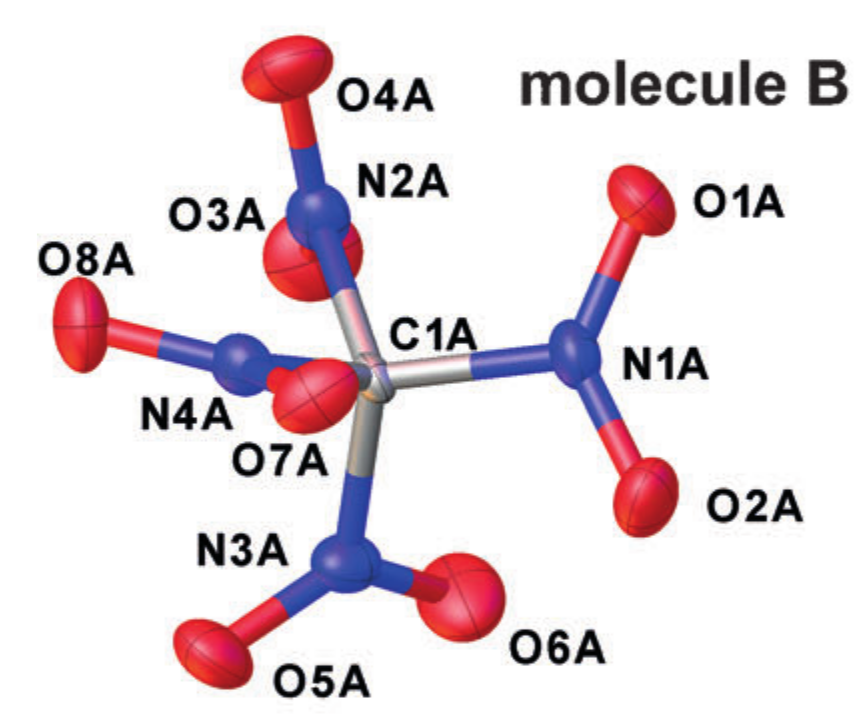
晶体学实例 r alpha 结构(晶体中为四硝基甲烷)。 改编自DOI:10.1002 / anie.201704396
每个原子都由描述其振动运动的椭球近似,并将所得椭圆的中心之间的距离作为原子间距离。 但是,原子运动性质的这种简化对应于引入谐波振荡器近似振荡,并且它并不总是能很好地工作。
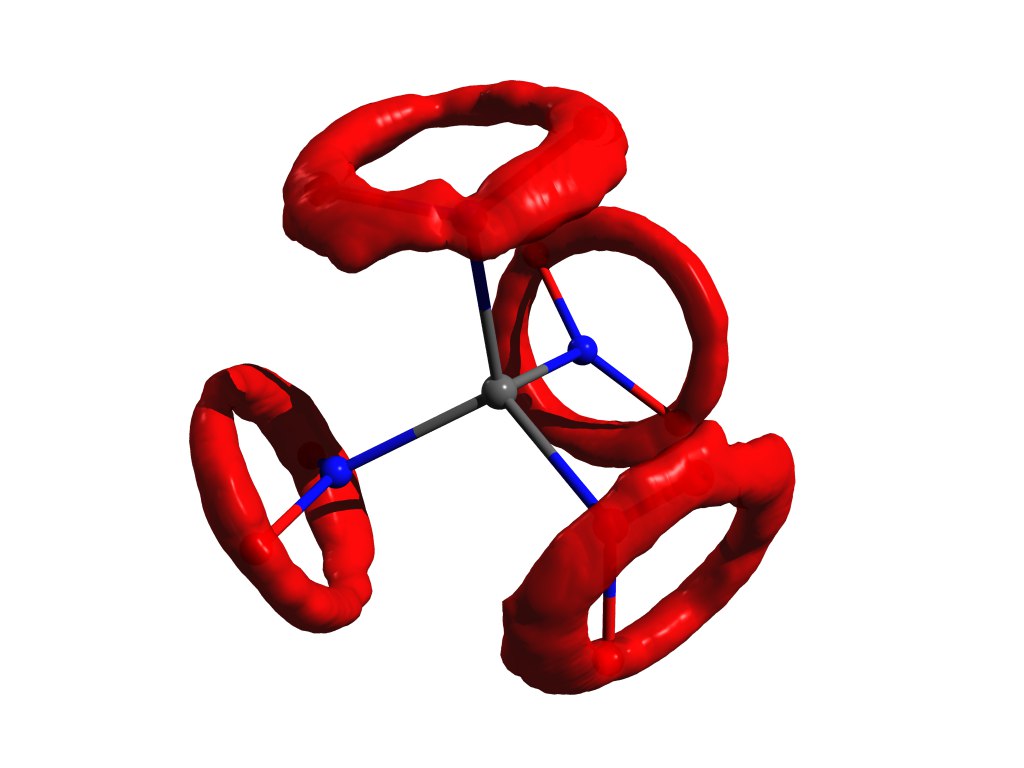
当谐波振荡器逼近不起作用时原子分布的一个示例。 与四硝基甲烷相同,但在气体中。
- rh1=??? 。 简略地描述了这种
十六进制奇迹柔道鱼鲸,它与现实之间的联系非常微弱,但是它具有奇妙的特性:必须在几何上保持一致(请参见下文),并且可以轻松地进行计算。 因此,她在电子照相界获得了极大的欢迎。
在微波光谱学中,结构变体略少。
- 身体上最容易理解的是 rn= langlen| hatr|n rangle 结构。 实际上,这是分子在一定振动状态下的平均几何形状( |n rangle ) 由于它们最常与冷分子一起工作,因此通常会观察到此类中的一种特定结构: r0 ,即 当原子围绕其最有利的位置进行零振动时,处于基振动状态的分子的几何形状。
- 最受欢迎的是 rs 结构。 下标“ s”表示“替代”。 他们以这种方式得到结果:他们相信原子在空间中固定有一些坐标,然后对分子中的原子进行单原子同位素取代,并通过改变旋转常数来确定该原子的位置。 该技术的主要优点是简单。 减号:您只需要单取代+并非可以像这样建立所有原子位置+这种模型的物理含义不是很清楚。
- 逻辑发展 rs 结构是 rm -通过质量加权最小二乘拟合获得的结构。 他们还需要同位素取代的分子,但是任何一个都已经适合。
这远非所有可能的结构类型...
但是,“
伟大的模拟器”使用“ Opt”之类的魔术咒语时,使用某些标准量子化学软件包(例如
Gaussian Evil Corporation Program )的用户会得到所谓的“平衡几何”,或者
re 结构。 这是原子核的最佳配置,可将系统的电子能量降至最低。 而且,这种结构也可以脱离电子衍射和旋转光谱学,但仅适用于非常小的对称分子,并且可以与其他研究方法结合使用。 到目前为止,它还没有解决。
因此出现了一个问题:比较是否正确
re 具有一些实验性的结构,只看实验中的错误?
答案很简单:
不 ,有必要在可能的系统差异上附加一个错误。 对此可以给出一个非常生动的例子:Bastiansen-Morino效应(请参见文章
DOI:10.1107 / S0365110060002557和
DOI:10.1107 / S0365110060002545 )。
假设我们有一个分子类型为CX
2的分子(即CO
2 ,CS
2等)。 从学校化学过程中应该知道,这些分子具有线性结构(碳原子和两个X硫属元素位于一条直线上)。
这意味着X原子之间的距离应等于C-X键长度的两倍(即
re( mathrmXX)=2re( mathrmCX) )

无论如何,如果我们测量原子C和X之间的距离(
rg( mathrmCX) )和XX(
rg( mathrmXX) )通过气体电子衍射,我们得到
rg( mathrmXX)<2rg( mathrmCX) ,即 该分子证明是弯曲的。 原因在于该分子做所谓的
剪刀式振动 ,由于这种
振动 ,X原子彼此之间的距离比最有利的位置小得多(请参见下图)。

巴斯坦森-莫里诺效应从何而来。 图片来自
DOI文章
:10.1039 / C6CP05849C 。
因此,如果我们将温度平均值
rg 结构平衡
re ),我们会得出错误的结论,即二氧化碳和二硫化碳的分子是弯曲的。
这就是为什么在比较不同类型的几何参数时,必须始终非常小心的原因。 这既适用于它们之间的实验数据比较,也适用于实验和理论的比较。
分子标准模型分子
现在,让我们想象一下,我们全心全意地希望在理论模型的基础上模拟某些实验的结果,以便在公平竞争中将模拟与现实进行比较。
在这里还必须要小心,因为 不同模型的分子也有其适用范围。 让我们使用标准分子模型的示例对此进行研究。
首先,您需要了解什么是分子标准模型。 BAK物理学家有自己的
标准模型 ,天文学家有
自己的 模型 ,物理学家有自己的基本设计,以后可以从中进行跳舞。 但是与物理模型不同,我们考虑的是一组近似值,使用户可以相对自动和快速地获得结果。
对于高斯用户现在我们回想起高斯魔术法术“ Opt”和“ Freq”的基础。
引入的近似值的一般方案如下所示:
质量的最底层是我们的标准模型。 简短地经历其接收的所有阶段。
生成的模型称为RR-HO(@BO)。我们不会碰到Born-Oppenheimer(BO)近似,但是我们将不得不在结构化学的框架内谈论硬转子和谐波振荡器。这个近似的主要问题是分子不是刚性的,并且其振动是完全谐波的。因此,实际上,我们需要近似非刚性转子和非谐振荡器。这里的关键词是“非谐”,即“不是谐波。”让我们谈谈最简单的分子:双原子。它们有很多示例:HCl,HBr,HI,CO,O 2,N 2等。等它们与所有分子的区别在于它们只有一种振动:原子间距离的扩展/压缩。这是我们可以在气体电子衍射中测量的原子之间的距离(在平均温度的变化范围内, rg )和旋转光谱(平均而言,例如地面振动状态,即 r0 )
现在,主要的问题出现在生命的宇宙和一般的宇宙中:将会是什么 rg 和 r0 谐波振荡器的近似值,以及它与平衡距离的比较 re ?
要获得答案,您必须查看双原子分子的势能表面:
- 如果我们在谐波振荡器的逼近下工作,那么我们的电势将相对于平衡位置对称:向右或向左移动看起来相同。因此,分子在振荡过程中会以相同的频率左右移动,因此,平衡位置总是变成我们的平均位置。
- , , 2 :
- ( r→0 ), - , « » ,
- ( r→+∞ )
, , , , . , , . , ( re<r0,rg ), : 0.01 Å, .
因此,即使我们想要计算的实验更像是在标准分子模型(RR-HO @ BO)的框架内,也不会得到任何新的东西,因此,非常平衡的几何体将参与比较。错误结论2
 来自DOI文章的插图:10.1002 / anie.201611308。结论非常简单,由两部分组成。
来自DOI文章的插图:10.1002 / anie.201611308。结论非常简单,由两部分组成。- 通过正确的比较,所有值应具有相同的含义。
- 如果值不同,则不应忘记此值。
科学论文中的错误示例
《印度作品》
实际上,您可以找到的主要地方是低级杂志。它们很少包含具有出色结果的文章,因此被“领先研究者”从第二个或更多世界的国家(金砖国家(BRICS)国家及其不太成功的追随者)中选出。这里的“低级”杂志并不是指那些发表诸如“ The Rooter:接入点和冗余的典型统一算法”的文章,而是受人尊敬的科学出版物。在我的科学领域,最著名的“半洗法”是:(还有其他)。如您所见,根据正式迹象,在俄罗斯科学中,它们被认为是非常不错的出版物。但是,大量可疑质量的r涌入,仍然大量泄漏。作为说明,我阅读了最新一期的《分子结构杂志》,并浏览了目录,瞧:S. Sathiya,M。Senthilkumar,C。Ramachandra Raja,晶体生长,Hirshfeld表面分析,DFT研究和硫脲4二甲基氨基苯甲醛的三阶NLO研究。Struct。,V.1180(2019),PP。81-88。
https://doi.org/10.1016/j.molstruc.2018.11.067
此类工作的总体结构非常朴实。- 一些物质被“煮沸了”(但更经常是在Sigma上愚蠢地购买)。在这项工作中,该物质仍处于煮熟状态。
- (), . — , ( , ) , . , - Gaussian, « ». … 1 2 , .. - .
- , /Vis, .
- 在标准的分子可视化仪中,例如(粗糙的)GaussView,虽然可以绘制出精美的图片,但是始终质量不佳。
- 没有得出任何实质性的结论:“我们做了很多实验,计数了很多,带来了表格和图片 ⇒ 我们很棒,给我们糖果。”
但从我的最爱:文章M. Govindarajan,M。Karabacak,FT-IR,FT-拉曼光谱和紫外光谱研究;1-硝基萘的计算频率估计分析和电子结构计算// Spectrochimica Acta A Part:Molecular and Biomolecular Spectroscopy,V.85(2012),PP。251-260,https: //doi.org/10.1016/j.saa.2011.10.002
。
在其中,愚蠢地记录了固相中的光谱,然后根据气相HO模型中的平庸计算进行了解释。但诀窍在于,他们甚至无法进行常规计算,这是他们在文章评论中礼貌地暗示的。但是,“印度作品”一词(类似于“ 印度代码 ”一词)是指仅来自相应神秘次大陆的作品。如果您前往Cyberleninka的精彩站点,那么您会看到Abyss,而Abyss也会看着您,您会发现很多有趣的事情。搜索“量子化学»(C /无附加条件‘PCA’)能够找到很多东西魔鬼有用的。由于大量的“量子化学”著作专门用于真空中球形马的研究(即,不考虑实际情况的计算),因此它们与本文无关。但是其中有三件作品丢失了:我对后者特别满意,因为“充分评估”是对不同阶段(气体与晶体)和具有不同含义的结构的比较不足( re 与 rα )-这确实是足够的高度。好的杂志会发生这种情况吗?
是的,那里有“错误”。不要害怕,一切都顺利!, , , / . , , .
, : , .
我们正在谈论的是一种具有极长的C-C单键的有机分子:1,1'-bisdiadamantane:
为什么这个分子很酷,
(
), , C--C 1.54 Å.
, 1,1'-
re=1.630±0.005 Å, 0.08 Å ( )!
C--C - , , , , . , , () . - , . , ( ) , .
中央一元连接C-C很长,因此比较理论方法如何重现现实非常有趣。现实是在第一篇文章中,它们是:
天哪!— Nature, , — JACS — !!! , - .
仅由晶体学数据表示。结果,在不对参数差异进行适当校正的情况下,将比较困难的可比较事物彼此进行比较,他们最终得出结论,气体中中心C-C键的长度为1.655Å,超出0.02Å。这远远超过了实验误差。幸运的是,最后,他们与专家们就这些问题进行了合作,最终收到了正确的答案(此工作的简短摘要也可以在N +1上找到)。您需要比较吗?
 在我写完比较的正确性之后,可能会引起一个合理的问题:那么是否有必要将计算结果和实验结果之间进行比较?有必要!随您所需!有一个著名的说法(我找不到可靠的作者):
在我写完比较的正确性之后,可能会引起一个合理的问题:那么是否有必要将计算结果和实验结果之间进行比较?有必要!随您所需!有一个著名的说法(我找不到可靠的作者):除了进行理论计算的人之外,没有人相信。
除了获得实验结果的人,其他人都相信实验结果。
译成俄语,这听起来像:没人相信理论计算,只有进行计算的人相信,但是每个人都相信实验数据,除了接受实验的人。但是在科学中,每个人都必须相信(好吧,还是大多数),而实验是唯一的衡量标准,因为它将我们计算的结果与现实联系在一起。第二最重要的化学期刊上有一篇出色的文章(开放获取),主题是研究人员或阅读科学文章和/或新闻的普通人应该做什么:Mata R.,Suhm M. // Angew。化学诠释Ed。,56(2017),DOI:10.1002 / anie.201611308(顺便说一句,我已经给出了链接,因为里卡多·马塔(Ricardo Mata)的照片来自本文)。本文的结论为模拟理论家和实验者提供了建议。我将在这里给他们(翻译和稍作修订),作为这篇文章的最后一句话。- 理论家必须:
- 不仅给出成功的方法和尝试,还描述方法的失败(特别是如果这些方法很流行),
- 完整,完整地描述您的方法,
- 在可能的地方给出误差的估计(或描述),以及重要的可接受的近似值和简化值。
- 反过来,实验者必须:
- 戳理论家的实验数据,他们可以将其用作基准(标准),
- 以显示科学界难以理解的实验数据,其中的理论(或其他实验)将有助于做出解释,
- 用数据在
国外的理论会议上发言,
- 从实验数据中提取出尽可能多的东西,以便与理论进行比较,
所有最好和正确的比较! 请记住:只有无所事事的人才不会犯错。
聚苯乙烯
作为总结,我想提供一小部分数据库,您可以在其中挖掘分子的各种实验数据。结构数据库
结晶罐
确定晶体中分子结构的最简单方法是 PCA是例行程序。因此,如果您不知道分子的外观,请继续前往晶体学数据库(在这里收集几乎所有塞入测角计并被短波粒子束照射的物质的结构)。由于有很多这样的银行,所以我只列出最著名的银行(可以在Wiki上找到更完整的列表)。- 无机晶体结构数据库(ICSD)。这不完全是关于分子的,在那里您主要可以找到不同盐,金属,陶瓷等的结构。该基地得到了卡尔斯鲁夫技术大学的支持,因此使用该基地是付费的,而且价格不菲。但是,如果那样,她的网站。
- Cambridge Structural Database ( CSD ). , . ! . ! . , , , , . .
- Crystallography Open Database ( COD ). - . , , , , . .
- , , Protein Data Bank ( PDB ). , ( ). .
气体中分子的结构
这里的情况有些糟糕,因为 无论是进行(至少需要高真空)还是进行解释,研究自由分子结构的实验都非常复杂。因此,数据库少得多。- 最大的游离分子数据库是分子气相DOCumentation(或MOGADOC)。它位于乌尔姆大学,是一笔非常昂贵的投资。但是,如果有的话,该站点在这里。
- 如果您想知道分子的100%实验平衡结构,那么这就是NIST计算化学比较和基准数据库(CCCBDB)。几乎所有纯粹是实验性的re - , . .
- — MolWiki . , , , ( ). .
?
 摘自xkcd.com这里的数据库要多得多,因为无需解释就删除频谱比获得结构要简单得多(无需构建模型并证明它们是正确的)。此外,这些光谱具有重要的应用价值:它们可用于确定样品的成分,无论是来自邻近河流的水还是来自分子云的信号(甚至来自系外行星大气的信号,请参见上图)。是的,顺便说一下,本节中的所有链接都是免费数据库。
摘自xkcd.com这里的数据库要多得多,因为无需解释就删除频谱比获得结构要简单得多(无需构建模型并证明它们是正确的)。此外,这些光谱具有重要的应用价值:它们可用于确定样品的成分,无论是来自邻近河流的水还是来自分子云的信号(甚至来自系外行星大气的信号,请参见上图)。是的,顺便说一下,本节中的所有链接都是免费数据库。- NIST Chemistry WebBook . , . , UV/Vis, - . , ! , , . .
- High-resolution transmission molecular absorption database ( HITRAN ). - , , , , (, ). .
- , The Cologne Database for Molecular Spectroscopy, . .
- 好吧,作为最后一个示例,我将介绍ExoMol项目。这些不是完全纯的实验光谱,但这是理论与实验相互作用的一个很好的例子:基于高精度的实验数据和非常高级的计算,可以在不同(包括极端)条件下预测简单分子的光谱。这里的主要重点是生物标记,因此,当天文学家看到系外行星的光谱时,他们可以轻松地识别出我们已经在其中知道的分子。该网站。
PPS
如果有错误/仍然无法理解,请在评论中写-我会更正/尝试更好地解释。