Mnogabukaff认为,量子化学考虑了μ子催化的原理:μ子到底如何降低所需等离子体的温度。 分两部分(第一部分可以在
此处阅读)。
第二部分的本质很简单:μ子比电子重,因此它提供了更强的化学键和更接近的原子核,从而降低了引发热核反应所需的等离子体温度。
但是,那些希望查看分子式,图表并看到应用于最简单(准)分子的量子化学概念实质的人,受到了欢迎。
引言
在第一部分(请参阅
此处 )中,我们检查了氢原子之间的差异
mathrmH cdot= mathrmp+e− 从笨重的介子对应
mathrmp+ mu− :在第二种情况下,μ子将被更牢固地束缚,并与质子距离更近。 同时,我们研究了这里需要的一些重要事物(轨道的形式和单位的原子系统)。
在第二部分(即此处)中,我们将尝试理解为什么点燃热核反应所需的等离子体温度降低,降低程度和降低程度。 我们感兴趣的反应是:
mathrmnH+mH rightarrow text新内核+能量
其中n,m = 1,2,3分别对应于质子,氘和tri。 这些核自然会带正电荷,因此,如果您试图使其更靠近,它们将根据库仑定律(请参阅
上一部分 )开始排斥,这是防止聚变反应发生的非常大的障碍。 顺便说一句,在核衰变反应的情况下,这种排斥作用是相反的,因为在与共同核分离之后,彼此排斥的碎片获得了额外的动能,而正是这种能量在核电站中被加热。
为了克服这种库仑势垒,需要提高等离子体温度(
T ),正如大家从
MKT的学校课程中所记得的那样,通过以下公式将其与等离子体中的平均粒子速度(
v )相关联
mv2=3k mathrmBT
其中
m是粒子的质量,并且
k mathrmB -
玻耳兹曼常数 。
但是,让我们想象一下,我们已经将两个氢核组合成一个特定的粒子,它们已经位于附近,因此对它们的其余障碍已经很小。 然后,我们需要显着加速这些粒子(请阅读:我们需要更低的温度),以便将它们组合成新的事物。 而这样的作用应该起到中间离子的作用
( mathrmnH mu− mathrmmH)+ 氢分子离子的类似物
mathrmH+2=( mathrmHe−H)+ 。
在检查了这两个粒子之间的差异之后,我们将认识到μ子在降低热核聚变的着火温度方面的有效性。
牛奶 MO LKAO方法
因此,我们有一个分子系统,该系统由2个带电荷+
e的氢核(一个电子电荷模)和一个带电荷
e的粒子(电子或介子)组成。 我们的系统在与其他粒子碰撞之前一直处于隔离状态,因此可以将其能量分解为以下组成部分:
E=T( mathrmH1)+T( mathrmH2)+\括号T( mathrme−/ mu−)+V( mathrmH1 textfromH2)+V( mathrm mathrme−/ mu− text mathrmH1)+V( mathrm mathrme−/ mu−\文字k mathrmH2)E mathrme
前两个词(
T( mathrmH1) 和
T( mathrmH2) )是氢核的动能,第三项(
T( mathrme−/ mu−) )是负粒子(电子或介子)的动能,第四项
V( mathrmH1\文字fromH2) 是氢相互之间排斥氢的库仑能量,其余两个是电子/介子对每个质子的库仑吸引力。 在一般情况下,这是一个三体问题,只是一个量子问题。 自然地,解决额头上的问题非常困难。 但是,幸运的是,核比电子重至少1800倍,比介子重10倍,因此它们的运动明显比小的负粒子慢。 因此,您首先可以依次解决问题:首先,找到与原子核运动无关的运动能量,即
E mathrme 然后充满能量。 看起来像这样。
- 选择氢原子相对于彼此的排列,这决定了氢原子之间以及与电子/介子之间的库仑相互作用。 库仑势 V(R)=k fracq1q2R 仅取决于粒子电荷 qi 以及它们之间的距离,因此对于所有氢同位素而言,该值都将相同。 此外,解决了这些原子核领域中电子/介子的运动问题。 这是一个身体的任务。
- 这些能量 E mathrme 计算原子核相对于彼此的所有可能排列,这将是原子核运动的有效势能。 在我们的案例中,我们需要计算彼此之间不同距离处的能量,因此一对原子核的电势始终是一维的。 好吧,那么我们只需要解决两个氢同位素相对于彼此运动的两体问题。
显然,与我们有关的问题的根源是计算原子核领域中的电子/μ子能
E mathrme 。 实际上,这是化学键:在某些位置将核保持在一起的特定电势。 而寻找化学键能的这一任务是量子化学中的主要任务。
不幸的是,介子和电子都是量子粒子,因此,为了找到这种能量,我们必须求助于量子力学方法。 实际上,我们在两个相同原子核场中的电子/介子运动问题已得到明确解决(请参见
此处 ),但是该解决方案非常复杂,其结果不像氢原子那样清晰。 因此,我们将尝试分解适用于任何系统的其他近似方法。 这就是所谓的 分子轨道法作为原子轨道或MO LKAO的线性组合。
让我们仔细看一下氢原子核领域中电子/介子运动的薛定ding方程:
hatH psi= underbrace left( overbrace− frac12m( frac\部分2\部分x2+ frac\部分2\部分y2+ frac\部分2\部分z2)\帽子T+\大括号− frac1R1 hatV1+ overbrace− frac1R2 hatV2+ overbrace frac1R hatV mathrmHH right) hatH psi=E psi
该方程式是用原子单位制写的(请参见
前面的PS ),因此,氢核的电荷和电子/μ子的电荷分别为+1和-1,电子质量为
m = 1,且μ= 207。
如果仔细看一看,您会发现在哈密顿量中,您可以选择与负粒子仅围绕一个原子核运动的纯粹运动相关的片段,这仅是氢原子的哈密顿量,这可以通过两种方式完成:
hatH=( overbrace hatT+ hatV1 hatH1+ hatV2+ hatV mathrmHH)=( overbrace hatT+ hatV2 hatH2+ hatV1+ hatV mathrmHH)
类氢原子的哈密顿量之外(
hatHi, i=$1. ),我们总是有2个块:电子/介子与另一个原子核相互作用的能量(
\帽子Vj )和核排斥能(
\帽子V mathrmHH ) 它们中的第二个根本不影响电子的运动-只是一定程度的能量转移,但是电子与另一个原子核的相互作用很重要。
我们可以想象,在任何时刻,我们的粒子仅绕一个原子核旋转,而与第二个原子核的相互作用只是一个校正。 作为绕一个原子核旋转的一种方法,我们可以假定电子/介子处于基态(1s),其波函数从上一部分中为我们所熟知:
|1s rangle= frac1 sqrt pi exp left(− fracRR1 right)
在哪里
R1 是粒子的玻尔半径。 在电子的情况下
R1=1 硼(它是电子的玻尔半径,大约等于0.5埃),在μ子的情况下
R1= frac1m mu\大约 frac1207 。
为了以某种方式近似2核场中的电子/μ子波函数,我们可以尝试采用以下表示形式:
psi\大约c1|1s1 rangle+c2|1s2 rangle
然后用我们求解一个复杂的偏微分方程的问题简化为寻找2个未知系数
c 1和
c 2 。 这是一个非常分子的轨道,以系数(科学的线性组合)原子1s轨道的总和表示。
自然,我们需要这些参数的方程式。 如果在Schrödinger方程中代入该近似值,则得到它非常简单
\帽子H psi=E psi :
hatH(c1|1s1 rangle+c2|1s2 rangle)=c1 hatH|1s1 rangle+c2 hatH|1s2 rangle=E(c1|1s1 rangle+c2|1s2 rangle)=c1E|1s1 rangle+c2E|1s2 rangle
实际上,我们希望这个比率在任何地方都可以满足,因此我们可以通过某种方式计算出这一切的平均值。 我们将左边的等式乘以
<1s1| 和
<1s2| 并整合所有坐标。 结果,我们获得了一个由2个线性方程组成的系统,其中必须找到系数
c 1 ,
c 2和能量
E :
beginpmatrix langle1s1| hatH|1s1 rangle& langle1s1| hatH|1s2 rangle langle1s2| hatH|1s1 rangle& langle1s2|\帽子H|1s2 rangle\结束pmatrix\开始pmatrixc1c2 endpmatrix=E beginpmatrix langle1s1|1s1 rangle& langle1s1|1s2 rangle langle1s2|1s1 rangle& langle1s2|1s2 rangle endpmatrix beginpmatrixc1c2 endpmatrix
学习过线性代数的任何人都会认识到广义特征向量-特征值问题。 在解决它之前,我们将分析两个矩阵(现有的tuta)的元素相等(并同时用一个字母引入它们的简称)。
- 让我们从最简单的开始: langle1s1|1s1 rangle= langle1s2|1s2 rangle=1 -这是波函数的归一化,正如我们回想的,在某处找到电子/μ子的总概率至少为1。
- langle1s1|1s2 rangle= langle1s2|1s1 rangle=S -这就是所谓的 重叠积分,显示1s电子云如何与每个原子重叠。
- langle1s1| hatH|1s1 rangle= langle1s2| hatH|1s2 rangle= alpha 。 该积分包括几个部分:
langle1s1| hatH|1s1 rangle=\底线 langle1s1| hatH1|1s1 rangle− fracm2+ langle1s1| hatV2|1s1 rangle+ frac1R
- langle1s1| hatH|1s2 rangle= langle1s2| hatH|1s1 rangle= beta 。 这里是类似的:
\ langle 1s_2 | \ hat {H} | 1s_1 \ rangle = \ underbrace {\ langle 1s_2 | \ overbrace {\ hat {H} _1 | 1s_1 \ rangle} ^ {-\ frac {m} {2} | 1s_1 \ rangle}} {{\ frac {m} {2} S} + \ langle 1s_2 | \帽子{V} _2 | 1s_1 \ rangle + \ frac {S} {R}
即 类氢原子和核间斥力的能量,由重叠积分(第一项和最后一项)定标,以及电子/介子从一个原子跳到另一个原子的能量。
让我们从重写为的方程式中找到类氢离子能量的表达式
\开始pmatrix alpha& beta beta& alpha\结束pmatrix\开始pmatrixs1c2\结束pmatrix=E\开始pmatrix1&SS&1\结束pmatrix\开始pmatrixc1c2\结束pmatrix
要找到能量,您需要解方程:
det beginpmatrix alpha−E& beta−ES beta−ES& alpha−E endpmatrix=( alpha−E)2−( beta−ES)2=0
其中“ det”表示行列式(矩阵的行列式,俄语)。
关于
E的该二次方程式的解是:
E pm= frac alpha pm beta1 pmS=− fracm2+ frac1R+ frac langle1s1| hatV2|1s1 rangle pm langle1s2| hatV2|1s1 rangle1 pmS
第一块显然是原子的能量,第二块是核之间的排斥力,即阻止热核反应点火的同一个库仑势垒,必须处理最后一个复杂的结构。
如果我们放弃核间排斥力,而核间排斥力只是电子/μ子能级的参考点,我们会得到两个带能态
epsilon pm=− fracm2+ frac langle1s1| hatV2|1s1 rangle pm langle1s2| hatV2|1s1 rangle1 pmS
由于两个波函数
|1s1 rangle 和
|1s2 rangle -正面,以及
\帽子Vi<0 (因为负粒子总是被吸引到正粒子),然后
epsilon+<− fracm2 (单个原子的能量),以及
epsilon−>− fracm2 ,即 我们得到了分子轨道的标准图:
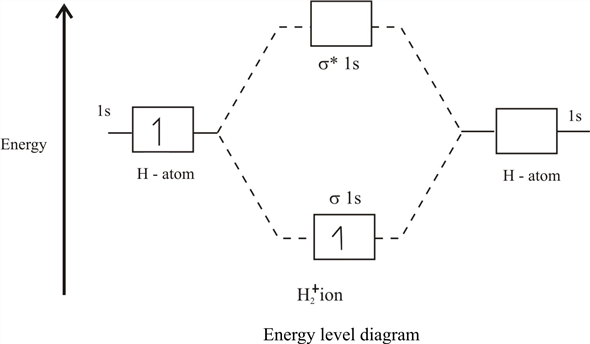
较低轨道的能量
E+ 称为绑定,顶部(具有能量
E− )-防粘连或松动。 结果,如果一个电子/介子坐在较低的分子轨道上,那么它从绕2个核飞行而不是绕1个核中受益,随着它的运动,它降低了系统的总能量。 这是一个非常神奇的化学键,可以屏蔽核之间的排斥,使核彼此相邻相当长的一段时间。
在这里,必须计算化学键的积分,以了解氢核的允许距离。 实际上,所有这三个受欢迎的积分都是经过分析计算的,但它非常痔疮且复杂(任何感兴趣的人,请参阅
Flary的《量子化学》一书的第9章)。 因此,我们将采用一种更简单的方法,并使用蒙特卡洛方法以数值方式计算这些积分。
都市法
在热核能的文字中,我向她的祖父表示敬意是非常合乎逻辑的:军事原子,更具体地说是
曼哈顿计划 。 正是从他那里产生了蒙特卡罗方法,尤其
是Metropolis算法 ,其中的作者之一Edward Edwarder是“氢弹之父”(即在Envetok环礁上发起热核聚变的人)。
通常,我们将分析该方法的本质。 它旨在用于统计力学的任务。 其中的主要分布是Boltzmann分布:在特定状态下检测到系统的概率为
exp(− betaE) ,
beta−1=k mathrmBT 。 并且系统在热力学平衡中某些参数
A的观测值等于积分
langleA rangle= frac1Z intA(q) exp(− betaE(q))dq
其中
q是参数化系统状态的坐标(例如,粒子的坐标/矩),
Z是归一化因子,称为分区函数:
Z= int exp(− betaE(q))dq
如果系统中有很多粒子,那么不计算额头中的任何积分都是完全不现实的。 如果系统的确实存在可能发生概率的状态,则朴素的蒙特卡洛方法(其中我们仅选择一堆随机
q坐标)也不会给出任何有意义的信息
exp(− betaE) 明显非零,很少。 正是在这种情况下,我们需要按重要性进行采样,在这种情况下,我们允许算法仅对状态空间中足够可能的位置进行采样。
Metropolis算法如下所示。
- 启动仿真时,我们在配置空间中选择一些起始近似值 mathbfq(0) 和一些最大可能增量的向量 delta mathbfq 。 首先,我们计算系统的能量 E ^ {{(0)} = E(\ mathbf {q} ^ {{0)}) (已读-概率 p= exp(− betaE(0)) )
- 第n步的新配置如下。
- 计算试用配置的能量 E mathrmtrial=E( mathbfq mathrmtrial) (即概率 p mathrmtrial= exp(− betaE mathrmtrial) )
- 然后我们比较旧的概率 p(n) 试用 p mathrmtrial
- 如果新配置具有更大或相同的可能性( fracp mathrmtrialpn geq1 ),或者等效地,新点的能量低于或等于旧点的能量( E mathrmtrial leqE(n) ),然后接受新点,系统进入该点( q(n+1)=q mathrmtrial ),
- 如果试验配置的能量较高( E mathrmtrial>E(n) ),相当于 fracp mathrmtrialp(n)<1 ,那么在这种情况下,我们生成一个随机数 P in[0;1) 从均匀分布中提取出来,并将其与概率比(即转移概率)进行比较。 如果 P< fracp mathrmtrialp(n) ,那么我们接受一个新观点,如果不接受( P geq fracp mathrmtrialpn ),然后我们拒绝,系统仍保持旧配置( q(n+1)=q(n) )...
- 根据上述算法采取许多步骤,我们对系统配置可能的空间中的重要部分(即非常重要的部分)进行了采样。 我们感兴趣的积分由以下公式计算:
langleA rangle= frac1Z intA( mathbfq) exp(− betaE( mathbfq))d mathbfq= frac1N sumNn=0A( mathbfqn)
这就是Metropolis算法的工作方式。
现在,有必要使它适应于我们感兴趣的3个积分的计算。 让我们更详细地了解它们。
- S(R)= langle1s2|1s1 rangle= int\极限+ infty− infty int\极限+ infty− infty int\极限+ infty− infty overbrace underbrace frac1 sqrt pi exp(−m underbrace| mathbfr− mathbfr2|R2)1s2A( mathbfr) cdot overbrace underbrace frac1 sqrt pi exp(−m\底线| mathbfr− mathbfr1|R1)1s1p( mathbfr)dxdydz 在哪里 mathbfr=(x,y,z) mathbfT -电子/μ子的坐标, mathbfri=(xi,yi,zi) mathbfT 是氢核的坐标,并且 Ri=| mathbfr− mathbfri|= sqrt(x−xi)2+(y−yi)2+(z−zi)2 -正负粒子之间的距离,
- langle1s1| hatV2|1s1 rangle=− int\极限+ infty− infty int\极限+ infty− infty int\限制+ infty− infty overbrace underbrace frac1 sqrt pi exp(−mR1)1s1 frac1R2A( mathbfr) cdot overbrace underbrace frac1 sqrt pi exp(−mR1)1s1p( mathbfr)dxdydz
- langle1s2| hatV2|1s1 rangle=− int\极限+ infty− infty int\极限+ infty− infty int\限制+ infty− infty overbrace underbrace frac1 sqrt pi exp(−mR2)1s2 frac1R1A( mathbfr) cdot overbrace underbrace frac1 sqrt pi exp(−mR1)1s1p( mathbfr)dxdydz
可以看出,如果我们针对概率
p计算原子之一的1s函数,
这样做当然不是很好,因为概率密度是平方波函数的模数 | psi|2 ,而不是wave函数本身 psi 。
那么在积分符号下的所有其他函数(第二个波函数以及在3种情况中的2种中,电子/μ子吸引原子核的电势)将是一个其平均值被计算的函数。 与通过Metropolis方法进行的通常计算相比,唯一要做的是拉直积分的归一化。 事实是标准规范化将继续进行
Z= int\极限+ infty− infty int\极限+ infty− infty int\极限+ infty− infty exp(−mR)dxdydz=4 pi int\极限+ infty0 exp(−mR)R2dR= frac8 pim3
我们需要规范化
sqrt langle1s1|1s1 rangle 在哪里
langle1s1|1s1 rangle= int\极限+ infty− infty int\极限+ infty− infty int\极限+ infty− infty exp(−2mR)dxdydz=4 pi int limits+ infty0 exp(−2mR)R2dR= frac pim3
这意味着根据都市计算的每个积分都需要乘以一个因子
fracZ sqrt langle1s1|1s1 rangle=8 sqrt frac pim3
这可以已经以某种脚本的形式进行了安排,例如在Python中(例如,下面的代码)。
像这样import numpy as np from math import *
使用这种计算,我们最终可以比较氢离子中的势能
mathrmH+2 和它的μon对应物。
mathrmH+2=p+e−p+ 与 米一吨ħ ř 米p +米Ü - p +
因此,借助脚本,我们可以计算由电子和介子束缚的氢核接近的势能表面。 作为能量参考点,我们将原子彼此无限稀释(即,
- 米/ 2 ,它等于原子核之间距离的电势
R = + i n f t y )
对于电子,接近最小值的电势如下所示:

最小值发生在大约2硼的距离处(即大约2个原子半径的总和),并且分子分解成碎片的能量大约为0.06 Hartree,这相当于加热到大约20,000开氏度(或摄氏温度,在这里无关紧要)。 为了转换能量,我建议使用诸如
this的在线资源。
离子键结合的氢离子的类似情况:

由于μ子氢的玻尔半径较小(请参见
上一部分 ),因此在最小势能下,氢核的距离也接近200倍。 该分子的分解能已经超过10 Hartree,相当于温度超过3 lyam度(
\大约( 3.2 ç d ö 吨10 6 ) Ç 我ř Ç )
为了点火,反应通常需要大约
10 8 K的温度,大约是320 Hartree。 让我们看看在普通的divodoron离子的情况下以及在其μ子形式的情况下,在多远的距离处都能获得相似的能量:
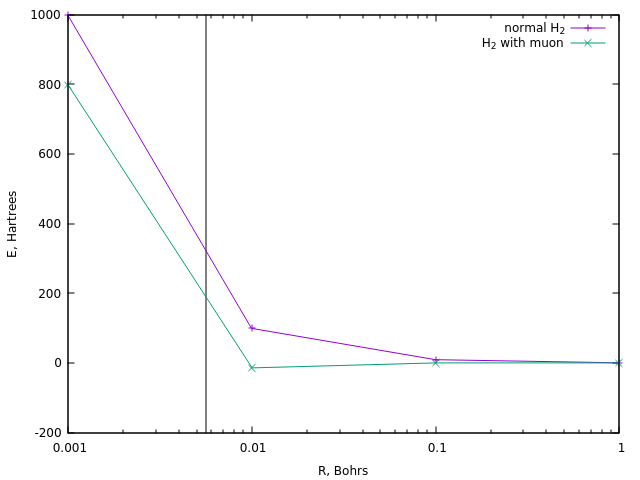
在前者的情况下,这相当于约0.0058硼(垂直线)的距离。
在约190 Ha的能量下,即在μon氢中,实现了类似的距离。 大约少一倍半。 这是对μ子催化温度的最简单估计。
但实际上,一切都会变得更酷。 事实是,如果形成稳定的颗粒
中号一吨ħ ř 米(米 ħ (米Ù - ) n H ) + ,那么在μ子活着时,这些原子核将相对彼此振荡。 从“两个氢原子”状态到“重核”状态的隧穿可能会发生,隧穿的可能性取决于所需的隧穿长度
d p - d ,因此通过μ子使两个原子核更靠近,我们将大大增加该反应隧穿过程的可能性。 不幸的是,对此效应的估计不再需要量子化学,而需要核物理学,因此,本部分讨论超出了本文的范围。 因此,在此我们将停止。
PS为什么不是那么简单?
实际上,在等离子体条件下形成这些颗粒并不是那么简单。 事实是,如果我们碰撞两个粒子,那么它们的总能量显然超过了离解能(在原子核+电子/介子的情况下为电离能),因此当它们碰撞时,它们不会形成稳定的粒子(原子,离子,分子),而是通过彼此过去。 为了相互坚持,他们需要抛弃一些多余的能量,为此,我们需要额外的第三个人来承担这种能量。 它可以是光子,也可以是某种在附近飞行的剩余粒子,但是最主要的是条件应该有助于这种过量能量的夹带。
PPS
如果您有任何评论/说明/问题,请在评论中或PM中写。 我将纠正所有问题,我将回答并解释所有问题。