如果您有一个清凉,健康的蛋白质想法,并且想将其变为现实该怎么办? 例如,您是否想通过创建一种结合了
大肠杆菌鞭毛蛋白片段和普通
幽门螺杆菌鞭毛蛋白的杂交蛋白来制造针对
幽门螺杆菌的疫苗(像
iGEM 2008的
斯洛文尼亚研究小组一样)?
斯洛文尼亚团队在iGEM 2008上展示的幽门螺杆菌杂种鞭毛设计出乎意料的是,由于基因组学,合成生物学以及云实验室的最新发展,我们几乎可以在不离开Jupyter笔记本的情况下创建所需的任何蛋白质。
在本文中,我将展示从蛋白质的概念到在细菌细胞中表达的Python代码,而无需接触移液管或与任何人交谈。 总成本只有几百美元! 使用
来自A16Z的Vijaya Pande的
术语 ,这是Biology 2.0。
更具体地说,在本文中,云实验室的Python代码执行以下操作:
- 编码我想要的任何蛋白质的DNA序列的合成 。
- 将该合成DNA 克隆到可以表达它的载体中。
- 用该载体 转化细菌并确认表达正在发生。
Python设置
首先,任何Jupyter记事本都需要常规的Python设置。 我们导入一些有用的Python模块并创建一些实用程序功能,主要用于数据可视化。
代号import re import json import logging import requests import itertools import numpy as np import seaborn as sns import pandas as pd import matplotlib as mpl import matplotlib.pyplot as plt from io import StringIO from pprint import pprint from Bio.Seq import Seq from Bio.Alphabet import generic_dna from IPython.display import display, Image, HTML, SVG def uprint(astr): print(astr + "\n" + "-"*len(astr)) def show_html(astr): return display(HTML('{}'.format(astr))) def show_svg(astr, w=1000, h=1000): SVG_HEAD = '''<?xml version="1.0" standalone="no"?><!DOCTYPE svg PUBLIC "-//W3C//DTD SVG 1.1//EN" "http://www.w3.org/Graphics/SVG/1.1/DTD/svg11.dtd">''' SVG_START = '''<svg viewBox="0 0 {w:} {h:}" version="1.1" xmlns="http://www.w3.org/2000/svg" xmlns:xlink= "http://www.w3.org/1999/xlink">''' return display(SVG(SVG_HEAD + SVG_START.format(w=w, h=h) + astr + '</svg>')) def table_print(rows, header=True): html = ["<table>"] html_row = "</td><td>".join(k for k in rows[0]) html.append("<tr style='font-weight:{}'><td>{}</td></tr>".format('bold' if header is True else 'normal', html_row)) for row in rows[1:]: html_row = "</td><td>".join(row) html.append("<tr style='font-family:monospace;'><td>{:}</td></tr>".format(html_row)) html.append("</table>") show_html(''.join(html)) def clean_seq(dna): dna = re.sub("\s","",dna) assert all(nt in "ACGTN" for nt in dna) return Seq(dna, generic_dna) def clean_aas(aas): aas = re.sub("\s","",aas) assert all(aa in "ACDEFGHIKLMNPQRSTVWY*" for aa in aas) return aas def Images(images, header=None, width="100%"):
云实验室
像AWS或任何计算云一样,云实验室拥有分子生物学设备以及通过Internet租借的机器人。 您可以通过单击界面上的几个按钮或编写自己编写的代码来向机器人发出指令。 不必编写自己的协议,就像我在这里将要做的那样,分子生物学的重要部分是标准的例行任务,因此通常最好依赖显示出与机器人良好交互作用的可靠的外来协议。
最近,出现了许多具有云实验室的公司:
Transcriptic ,
Autodesk Wet Lab Accelerator (测试版,并在Transcriptic的基础上构建),
Arcturus BioCloud (测试版),
Emerald Cloud Lab (测试版),
Synthego (尚未启动)。 甚至还有建立在云实验室之上的公司,例如专门研究CRISPR的
Desktop Genetics 。 有关在实际科学中使用云实验室的
科学文章开始出现。
在撰写本文时,只有Transcriptic属于公共领域,因此我们将使用它。 据我了解,大多数Transcriptic业务都建立在自动执行通用协议的基础上,因此用Python编写自己的协议(正如我将在本文中做的那样)并不常见。
 抄写式“工作单元”,底部配有冰箱,架子上配有各种实验室设备
抄写式“工作单元”,底部配有冰箱,架子上配有各种实验室设备我将在
自动协议中为Transcriptic机器人提供说明。 Autoprotocol是一种基于JSON的语言,用于为实验室机器人(和人类)编写协议。 自动协议主要在
此Python库上制作 。 该语言最初是创建的,现在仍受Transcriptic支持,但是据我所知,它是完全开放的。 有很好的
文档 。
一个有趣的想法是,您可以在自动协议上为中国或印度等偏远实验室的人员编写指令,并通过同时使用人员(他们的判断力)和机器人(缺乏判断力)获得一些好处。 我们需要在
这里提到
protocol.io ,这是对协议进行标准化以提高可重复性的尝试,但对于人类而非机器人而言。
"instructions": [ { "to": [ { "well": "water/0", "volume": "500.0:microliter" } ], "op": "provision", "resource_id": "rs17gmh5wafm5p" }, ... ]
自动协议片段示例分子生物学的Python设置
除了导入标准库外,我还需要一些特定的分子生物学工具。 此代码主要用于自动协议和转录。
“死体积”的概念通常可以在代码中找到。 这意味着转录机器人无法用移液管从试管中倒出最后一滴液体(因为他们看不到!)。 您必须花费大量时间来确保烧瓶中有足够的材料。
代号 import autoprotocol from autoprotocol import Unit from autoprotocol.container import Container from autoprotocol.protocol import Protocol from autoprotocol.protocol import Ref
DNA合成与合成生物学
尽管与现代合成生物学有联系,但DNA合成是一项相当古老的技术。 几十年来,我们已经能够制造寡核苷酸(即,长达200个碱基的DNA序列)。 但是,它总是很昂贵,化学反应也不允许长DNA序列。 最近,以合理的价格合成整个基因(多达数千个碱基)成为可能。 这项成就真正打开了“合成生物学”的时代。
克雷格·文特(Craig Venter)的
合成基因组学通过
合成整个有机体 (长度超过一百万个碱基),将合成生物学推向了最远。 随着DNA长度的增加,问题不再是合成,而是组装(即,将合成的DNA序列缝合在一起)。 对于每个装配,您可以将DNA长度增加一倍(或更多),因此经过十几次迭代后,您将获得一个
相当长的分子 ! 最终用户很快就会明白合成与组装之间的区别。
摩尔定律?
DNA合成的价格正在迅速下降,从一年前的0.30多美元跌至如今的0.10美元,但它的发展更像细菌而不是加工者。 相比之下,DNA测序价格的下降速度快于摩尔定律。 将
每个碱基0.02美元的目标
设置为拐点 ,您可以用简单的合成方法替换许多耗时的DNA操作。 例如,以这个价格,您可以
花费60美元合成一个完整的3kb质粒,而跳过一堆分子生物学。 我希望我们能够在几年内实现这一目标。
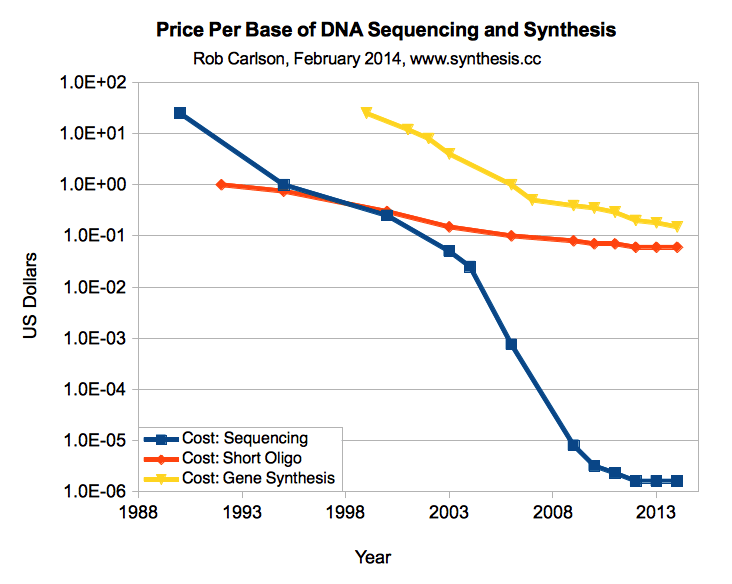 DNA合成价格与DNA测序价格的比较,以1个碱基为基础(Carlson,2014年)
DNA合成价格与DNA测序价格的比较,以1个碱基为基础(Carlson,2014年)DNA合成公司
DNA合成领域有几家大公司:IDT是寡核苷酸的最大生产商,也可以生产更长(最多2kb)的“基因片段”(
gBlocks )。
Gen9 ,
Twist和
DNA 2.0通常专门研究更长的DNA序列-这些是基因合成公司。 还有一些有趣的新公司,例如
寒武纪基因组学和
Genesis DNA ,正在研究下一代合成方法。
其他公司,例如
Amyris ,
Zymergen和
Ginkgo Bioworks ,也使用这些公司合成的DNA在人体上发挥作用。
合成基因组学也可以做到这一点,但是它可以合成DNA本身。
银杏最近
与Twist达成了一笔交易,获得1亿个基地:这是我见过的最大一笔交易。 事实证明,我们生活在未来,Twist甚至在Twitter上宣传了促销代码:当您购买1000万个DNA碱基(几乎是整个酵母基因组!)时,您还可以免费获得1000万个碱基。
 Twitter Twist利基报价
Twitter Twist利基报价第一部分:实验设计
绿色荧光蛋白
在此实验中,我们合成了简单的
绿色荧光蛋白 (GFP)的DNA序列。 GFP蛋白首先发现于在紫外线下发荧光的
水母中。 这是一种非常有用的蛋白质,因为它很容易仅通过测量荧光来检测其表达。 有可以产生黄色,红色,橙色和其他颜色的GFP选项。
有趣的是,各种突变如何影响蛋白质的颜色,这是一个潜在的有趣的机器学习问题。 最近,您将不得不在实验室中花费大量时间,但是现在我将向您展示,它(几乎)就像编辑文本文件一样容易!
从技术上讲,我的GFP是超级文件夹选件(sfGFP),具有某些突变以提高质量。
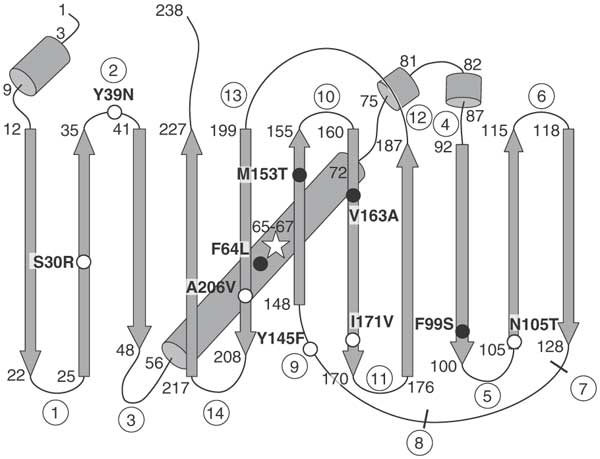 在superfolder-GFP (sfGFP)中,某些突变使其具有某些有用的特性。
在superfolder-GFP (sfGFP)中,某些突变使其具有某些有用的特性。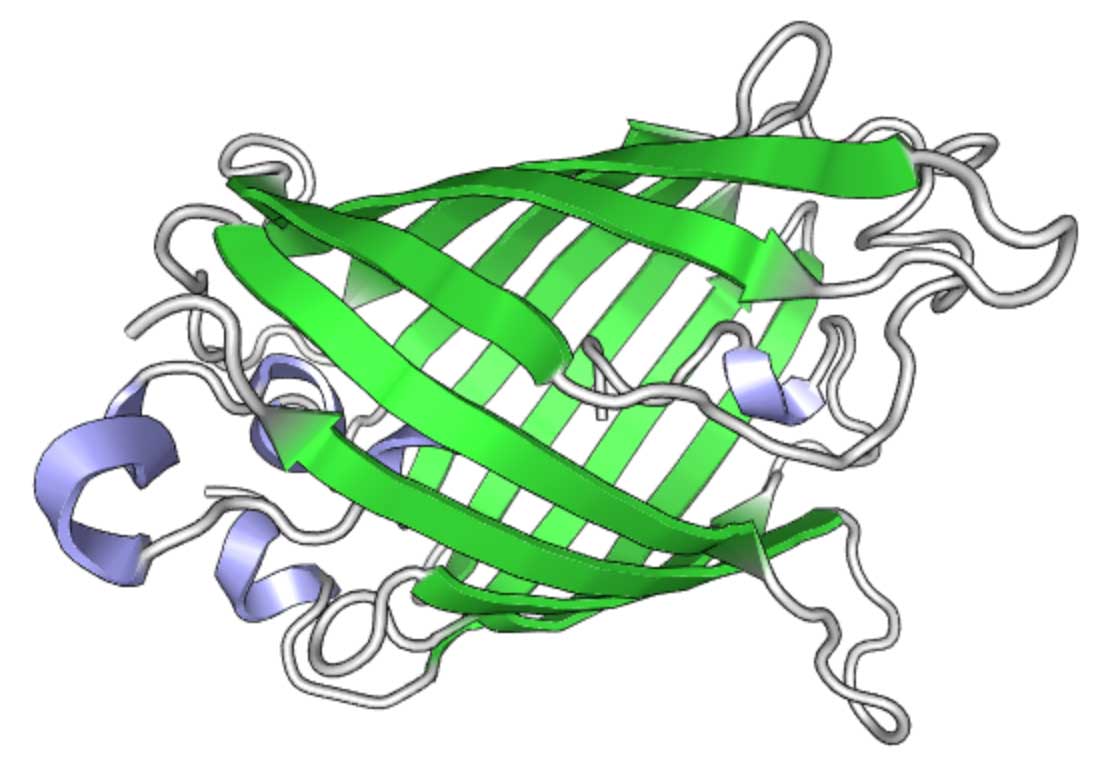 GFP结构(使用PV可视化)
GFP结构(使用PV可视化)Twist中的GFP合成
我很幸运地进入了Twist的alpha测试程序,所以我使用了他们的DNA合成服务(他们请了我的小订单-谢谢Twist!)。 这是我们领域中的一家新公司,具有新的简化合成过程。 它们的价格约为
每单位$ 0.10或更低 ,但是它们
仍处于beta版本 ,而我参与的alpha程序已关闭。 Twist筹集了大约1.5亿美元,因此他们的技术非常活跃。
我将我的DNA序列作为Excel电子表格发送给Twist(尚无API,但我想它将很快),他们将合成的DNA直接发送到了转录实验室的我的盒子中(我也使用IDT进行合成,但没有发送)转录本中的DNA,会稍微破坏乐趣)。
显然,此过程尚未成为典型的用例,需要一些支持,但它确实有效,因此整个管道仍然是虚拟的。 没有这个,我可能需要进入实验室-许多公司不会将DNA或试剂发送到他们的家庭住址。
 GFP是无害的,因此任何种类都可以突出显示
GFP是无害的,因此任何种类都可以突出显示质粒载体
为了在细菌中表达这种蛋白质,该基因需要生活在某个地方,否则编码该基因的合成DNA会立即降解。 通常,在分子生物学中,我们使用质粒,这是一种圆形DNA,位于细菌基因组之外,并表达蛋白质。 质粒是细菌共享有用的独立功能模块(例如抗生素抗性)的便捷方法。 一个细胞中可能有数百个质粒。
广泛使用的术语是质粒是
载体 ,合成DNA是插入(插入)。 因此,这里我们尝试将插入片段克隆到载体中,然后使用该载体转化细菌。
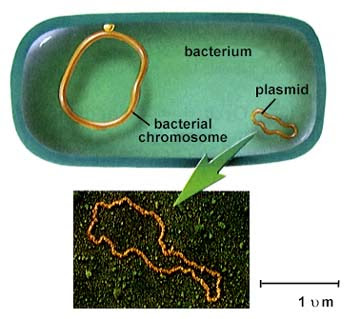 细菌基因组和质粒(不成比例!)( Wikipedia )
细菌基因组和质粒(不成比例!)( Wikipedia )pUC19
我在
pUC19系列中选择了一个相当标准的质粒。 该质粒非常常用,由于它是标准转录本清单的一部分,因此我们无需向其发送任何东西。
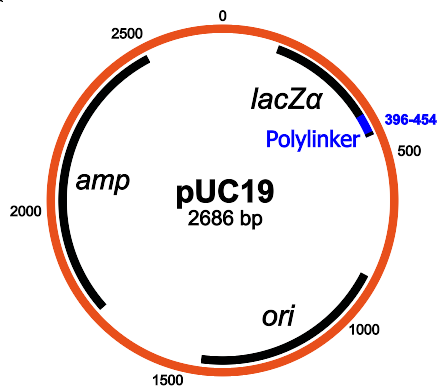 pUC19的结构:主要成分是氨苄青霉素抗性基因,lacZα,MCS /多接头和复制起点(维基百科)
pUC19的结构:主要成分是氨苄青霉素抗性基因,lacZα,MCS /多接头和复制起点(维基百科)PUC19具有很好的功能:由于它包含lacZα基因,因此您可以在其上使用
蓝白色选择方法,并查看插入成功的菌落。 需要两种化学品:
IPTG和
X-gal ,电路的工作方式如下:
- IPTG诱导lacZα表达。
- 如果通过在lacZα的多个克隆位点( MCS / polylinker )上插入的DNA使lacZα失活,则该质粒不能水解X-gal,这些菌落将是白色而不是蓝色。
- 因此,成功插入会产生白色菌落,插入失败会产生蓝色菌落。
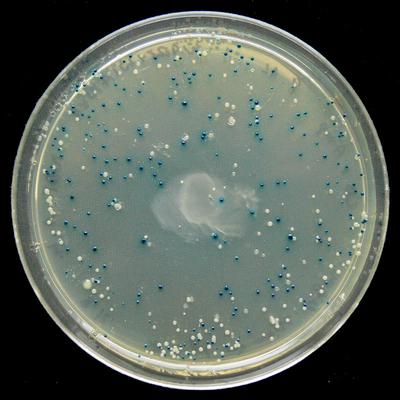 蓝色和白色选择显示lacZα表达被激活的地方( Wikipedia )openwetware文档
蓝色和白色选择显示lacZα表达被激活的地方( Wikipedia )openwetware文档说:
大肠杆菌DH5α不需要IPTG即可从lac启动子诱导表达,即使该菌株中表达了Lac阻遏物也是如此。 大多数质粒的拷贝数超过了细胞中阻遏物的数目。 如果需要最大表达量,请添加IPTG至终浓度1 mM。
合成DNA序列
SfGFP DNA序列
通过获取
蛋白质序列并
用适合宿主生物(此处为
大肠杆菌 )的
密码子对其进行编码 ,很容易获得sfGFP的DNA序列。 这是一种具有236个氨基酸的中等大小的蛋白质,因此10美分的DNA合成成本约为每个碱基
70美元。
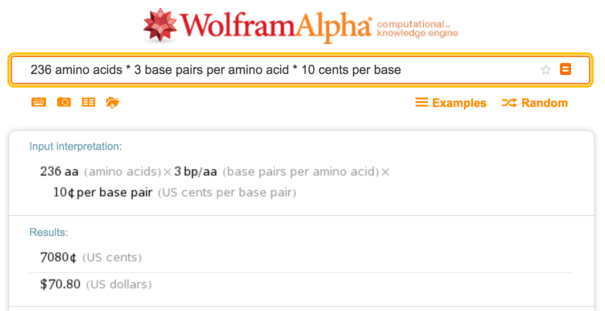 Wolfram Alpha,合成成本计算
Wolfram Alpha,合成成本计算我的sfGFP的前12个碱基是
Shine-Delgarno序列 ,我自己添加了该
序列 ,理论上应增加表达(AGGAGGACAGCT,然后ATG(
起始密码子 )起始蛋白质)。 根据
Salis Lab开发的计算工具(
演讲幻灯片 ),我们可以预期蛋白质的中等至高表达(翻译起始速率为10,000个“任意单位”)。
sfGFP_plus_SD = clean_seq(""" AGGAGGACAGCTATGTCGAAAGGAGAAGAACTGTTTACCGGTGTGGTTCCGATTCTGGTAGAACTGGA TGGGGACGTGAACGGCCATAAATTTAGCGTCCGTGGTGAGGGTGAAGGGGATGCCACAAATGGCAAAC TTACCCTTAAATTCATTTGCACTACCGGCAAGCTGCCGGTCCCTTGGCCGACCTTGGTCACCACACTG ACGTACGGGGTTCAGTGTTTTTCGCGTTATCCAGATCACATGAAACGCCATGACTTCTTCAAAAGCGC CATGCCCGAGGGCTATGTGCAGGAACGTACGATTAGCTTTAAAGATGACGGGACCTACAAAACCCGGG CAGAAGTGAAATTCGAGGGTGATACCCTGGTTAATCGCATTGAACTGAAGGGTATTGATTTCAAGGAA GATGGTAACATTCTCGGTCACAAATTAGAATACAACTTTAACAGTCATAACGTTTATATCACCGCCGA CAAACAGAAAAACGGTATCAAGGCGAATTTCAAAATCCGGCACAACGTGGAGGACGGGAGTGTACAAC TGGCCGACCATTACCAGCAGAACACACCGATCGGCGACGGCCCGGTGCTGCTCCCGGATAATCACTAT TTAAGCACCCAGTCAGTGCTGAGCAAAGATCCGAACGAAAAACGTGACCATATGGTGCTGCTGGAGTT CGTGACCGCCGCGGGCATTACCCATGGAATGGATGAACTGTATAAA""") print("Read in sfGFP plus Shine-Dalgarno: {} bases long".format(len(sfGFP_plus_SD))) sfGFP_aas = clean_aas("""MSKGEELFTGVVPILVELDGDVNGHKFSVRGEGEGDATNGKLTLKFICTTGKLPVPWPTLVTTLTYG VQCFSRYPDHMKRHDFFKSAMPEGYVQERTISFKDDGTYKTRAEVKFEGDTLVNRIELKGIDFKEDGNILGHKLEYNFNSHNVYITADKQKN GIKANFKIRHNVEDGSVQLADHYQQNTPIGDGPVLLPDNHYLSTQSVLSKDPNEKRDHMVLLEFVTAAGITHGMDELYK""") assert sfGFP_plus_SD[12:].translate() == sfGFP_aas print("Translation matches protein with accession 532528641")
读入sfGFP和Shine-Dalgarno:长726个碱基
翻译使蛋白质与登录号相匹配532528641
PUC19 DNA序列
首先,我检查
从NEB下载的
pUC19序列的长度正确,并且其中包含预期的
多接头 。
pUC19_fasta = !cat puc19fsa.txt pUC19_fwd = clean_seq(''.join(pUC19_fasta[1:])) pUC19_rev = pUC19_fwd.reverse_complement() assert all(nt in "ACGT" for nt in pUC19_fwd) assert len(pUC19_fwd) == 2686 pUC19_MCS = clean_seq("GAATTCGAGCTCGGTACCCGGGGATCCTCTAGAGTCGACCTGCAGGCATGCAAGCTT") print("Read in pUC19: {} bases long".format(len(pUC19_fwd))) assert pUC19_MCS in pUC19_fwd print("Found MCS/polylinker")
读入pUC19:2686个碱基长
找到MCS /聚接头
我们进行一些基本的质量控制,以确保EcoRI和BamHI仅在pUC19中存在一次(默认的转录本库存中提供以下限制酶):
PstI ,
PvuII ,
EcoRI ,
BamHI ,
BbsI ,
BsmBI )。
REs = {"EcoRI":"GAATTC", "BamHI":"GGATTC"} for rename, res in REs.items(): assert (pUC19_fwd.find(res) == pUC19_fwd.rfind(res) and pUC19_rev.find(res) == pUC19_rev.rfind(res)) assert (pUC19_fwd.find(res) == -1 or pUC19_rev.find(res) == -1 or pUC19_fwd.find(res) == len(pUC19_fwd) - pUC19_rev.find(res) - len(res)) print("Asserted restriction enzyme sites present only once: {}".format(REs.keys()))
现在,我们查看lacZα序列,并验证没有任何意外情况。 例如,它应该以Met开头,以终止密码子结尾。 通过将pUC19序列加载到免费的
snapgene浏览器中 ,也很容易确认这是完整的324bplacZαORF。
lacZ = pUC19_rev[2217:2541] print("lacZα sequence:\t{}".format(lacZ)) print("r_MCS sequence:\t{}".format(pUC19_MCS.reverse_complement())) lacZ_p = lacZ.translate() assert lacZ_p[0] == "M" and not "*" in lacZ_p[:-1] and lacZ_p[-1] == "*" assert pUC19_MCS.reverse_complement() in lacZ assert pUC19_MCS.reverse_complement() == pUC19_rev[2234:2291] print("Found MCS once in lacZ sequence")
lacZ的序列:ATGACCATGATTACGCCAAGCTTGCATGCCTGCAGGTCGACTCTAGAGGATCCCCGGGTACCGAGCTCGAATTCACTGGCCGTCGTTTTACAACGTCGTGACTGGGAAAACCCTGGCGTTACCCAACTTAATCGCCTTGCAGCACATCCCCCTTTCGCCAGCTGGCGTAATAGCGAAGAGGCCCGCACCGATCGCCCTTCCCAACAGTTGCGCAGCCTGAATGGCGAATGGCGCCTGATGCGGTATTTTCTCCTTACGCATCTGTGCGGTATTTCACACCGCATATGGTGCACTCTCAGTACAATCTGCTCTGATGCCGCATAG
r_MCS序列:AAGCTTGCATGCCTGCAGGTCGACTCTAGAGGATCCCCGGGTACCGAGCTCGAATTC
在lacZ序列中发现一次MCS
吉布森装配
组装DNA只是意味着交联片段。 通常,您将几个DNA片段收集到更长的片段中,然后将其克隆到质粒或基因组中。 在此实验中,我想将一个DNA片段克隆到lac启动子下方的pUC19质粒中,以便在
大肠杆菌中表达 。
克隆方法很多(例如
NEB ,
openwetware ,
addgene )。 在这里,我将使用Gibson程序集(
由Daniel Gibson在2009年的Synthetic Genomics中
开发 ),它不一定是最便宜的方法,而是简单而灵活的。 您只需要使用Gibson Assembly Master Mix将想要收集的DNA(有适当的重叠)放入试管中,它就会自行组装!
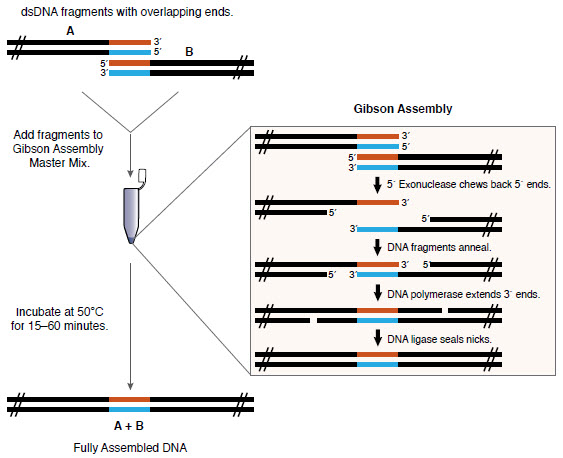 吉布森议会评论( NEB )
吉布森议会评论( NEB )原始资料
我们从在10μl液体中的100 ng合成DNA开始。 这等于0.21皮摩尔的DNA或10 ng /μl的浓度。
pmol_sfgfp = convert_ug_to_pmol(0.1, len(sfGFP_plus_SD)) print("Insert: 100ng of DNA of length {:4d} equals {:.2f} pmol".format(len(sfGFP_plus_SD), pmol_sfgfp))
插入:长度为726的100ng DNA等于0.21 pmol
根据
NEB组装协议 ,这已经足够了:
NEB建议在将1或2个片段组装到载体中时建议总共0.02-0.5皮摩尔的DNA片段,或者在收集4-6个片段时建议使用0.2-1.0皮摩尔的DNA片段。
0.02-0.5 pmoles * X微升
*优化的克隆效率是50-100 ng的载体,插入量超出2到3倍。 如果大小小于200 bps,则使用5次以上的插入。 吉布森组装反应中未过滤的PCR片段的总体积不应超过20%。
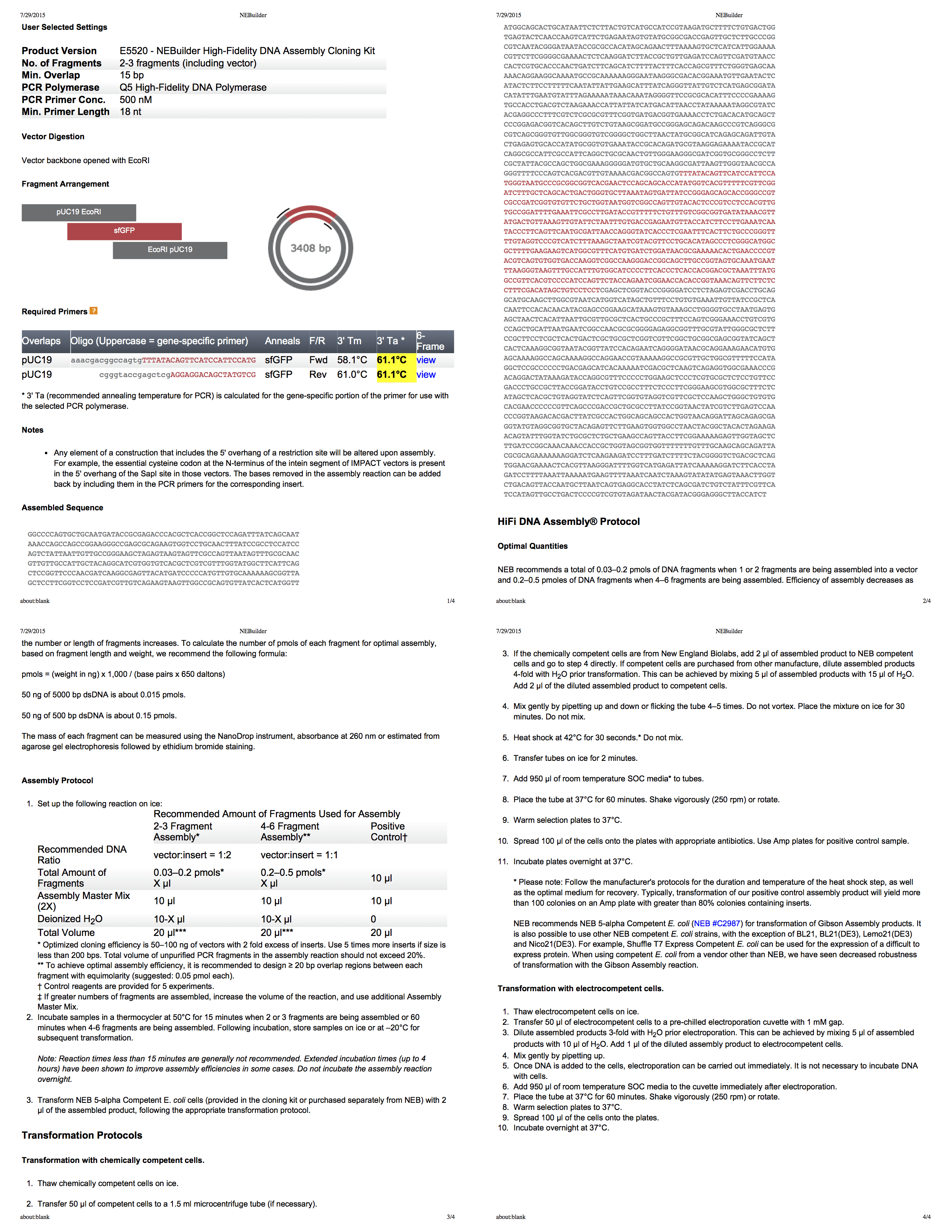
NEBuilder for Gibson组装
Biolab的NEBuilder是创建Gibson构建协议的绝佳工具。 它甚至可以为您生成包含所有信息的全面的四页PDF。 使用此工具,我们开发了用EcoRI切割pUC19的方案,然后使用PCR [PCR,聚合酶链反应可使生物材料中某些DNA片段的小浓度显着增加-大约。 per。]将适当大小的片段添加到插入中。
第二部分:实验
实验包括四个阶段:
- 聚合酶链插入反应以添加具有侧翼序列的材料。
- 切割质粒以适应插入。
- 通过Gibson插入和质粒组装。
- 使用组装的质粒转化细菌。
步骤1. PCR插入
吉布森的组装取决于您收集的DNA序列,该序列具有一些重叠序列(请参阅上面详细说明的NEB协议)。 除了简单的扩增,PCR还允许您通过在引物中简单地包含一个附加序列来添加侧翼DNA序列(也可以
仅使用OE-PCR进行克隆)。
我们根据上面的NEB协议合成引物。 我在Transcriptic网站上尝试
了Quickstart协议 ,但仍然有
一个auto- protocol 命令 。 转录本本身不合成寡核苷酸,因此在等待1-2天后,这些引物神奇地出现在我的清单中(请注意,引物的基因特异性部分在下面的大写字母中表示,但这只是装饰性的东西)。
insert_primers = ["aaacgacggccagtgTTTATACAGTTCATCCATTCCATG", "cgggtaccgagctcgAGGAGGACAGCTATGTCG"]
引物分析
您可以使用
IDT OligoAnalyzer分析这些引物的特性。
在调试PCR实验时,知道NEB方案将肯定会选择具有良好特性的引物,对了解引物二聚体的熔点和副作用的可能性很有用。 侧面的基因特定部分(大写)
熔体温度:51C,53.5C
全序列
熔体温度:64.5C,68.5C
发夹:-.4dG,-5dG
自二聚体:-9dG,-16dG
异二聚体:-6dG
PCR, , PCR. ( ), : . . , — .
代号 """ PCR overlap extension of sfGFP according to NEB protocol. v5: Use 3/10ths as much primer as the v4 protocol. v6: more complex touchdown pcr procedure. The Q5 temperature was probably too hot v7: more time at low temperature to allow gene-specific part to anneal v8: correct dNTP concentration, real touchdown """ p = Protocol()
WARNING:root:Low volume for well sfGFP 1 /sfGFP 1 : 2.0:microliter
sfGFP 1 /sfGFP 1 2.0:microliter {'dilution': '0.25ng/ul'}
sfgfp_pcroe_v5_puc19_primer1_10uM 75.0:microliter {}
sfgfp_pcroe_v5_puc19_primer2_10uM 75.0:microliter {}
Consolidated volume 52.0:microliter
Protocol 1. Amplify the insert (oligos previously synthesized)
---------------------------------------------------------------
✓ Protocol analyzed
11 instructions
8 containers
Total Cost: $32.18
Workcell Time: $4.32
Reagents & Consumables: $27.86 : PCR
在凝胶中,您可以在增加浓度(凝胶条在凝胶中的位置)和正确量(深色条)后评估产品的正确尺寸。凝胶具有对应于各种长度和数量的DNA的梯子,可用于比较。在下面的凝胶照片中,条带D1,E1,F1分别包含2μl,4μl和8μl扩增产物。与梯形图中的DNA相比,我可以估计每个泳道中的DNA数量(梯形图中每个泳道50 ng DNA)。我认为结果看起来非常干净。我尝试使用GelEval进行图像分析和浓度估算, , , , . . GelEval 40 /.
,
, dNTP , , 12,5 , 6 740bp 25 . GelEval 40 x 25 (1 2 ), , .
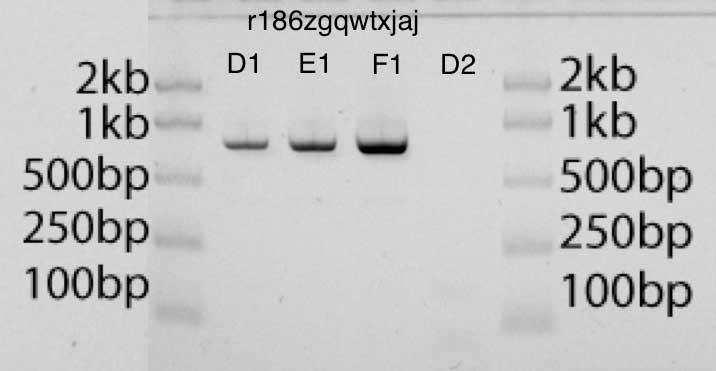 - EcoRI- pUC19, (D1, E1, F1), (D2)
- EcoRI- pUC19, (D1, E1, F1), (D2)PCR
Transcriptic最近开始从其机器人提供有趣且有用的诊断数据。在撰写本文时,它们尚无法下载,因此目前我只有热循环过程中的温度图像。数据看起来不错,没有意外的高峰或低谷。总共进行35个PCR循环,但是其中一些循环是在非常高的温度下进行的,作为PCR触地的一部分。在我之前尝试扩大这一细分市场的尝试中,有几种!-引物的杂交存在问题,因此这里的PCR在高温下工作了很多时间,应提高准确性。 用于着陆PCR的热循环诊断:封闭,采样和覆盖温度为35个循环和42分钟
用于着陆PCR的热循环诊断:封闭,采样和覆盖温度为35个循环和42分钟步骤2.切割质粒
sfGFP pUC19, . NEB,
EcoRI . Transcriptic , :
NEB EcoRI 10x CutSmart ,
NEB pUC19 .
, . , Transcriptic :
Item ID Amount Concentration Price
------------ ------ ------------- ----------------- ------
CutSmart 10x B7204S 5 ml 10 X $19.00
EcoRI R3101L 50,000 units 20,000 units/ml $225.00
pUC19 N3041L 250 µg 1,000 µg/ml $268.00
NEB:
. 10X dH2O 1X. , , , , . 50 5 10x NEBuffer , dH2O.
, 1 λ 1 37°C 50 . , 5-10 10-20 1- .
1 50 .
代号 """Protocol for cutting pUC19 with EcoRI.""" p = Protocol() experiment_name = "puc19_ecori_v3" options = {} inv = { 'water': "rs17gmh5wafm5p",
Volumes: re_tube:135.0:microliter water_tube:383.0:microliter EcoRI:30.0:microliter
Consolidated volume: 78.0:microliter
✓ Protocol analyzed
12 instructions
5 containers
Total Cost: $30.72
工作间时间:3.38美元
试剂和消耗品:$ 27.34
结果:质粒切割
我在略有不同的条件下和使用大小不同的凝胶进行了两次实验,但结果几乎相同。我都喜欢这两种凝胶。最初,我没有为“死”体积分配足够的空间(在1.5 ml的试管中,死体积为15μl!)。我认为这可以解释D1和E1之间的区别(两个频段应该相同)。通过在协议开始时创建稀释的EcoRI的正常工作供应量,可以轻松解决死体积问题。尽管存在此错误,但在两种凝胶中,D1和E1谱带在2.6kb的正确位置均具有很强的谱带。在条带D2上,未切割的质粒:如预期的那样,它在一种凝胶中不可见,而在另一种凝胶中几乎不可见。两张凝胶照片看起来完全不同。部分原因是该步骤Transcriptic尚未自动化。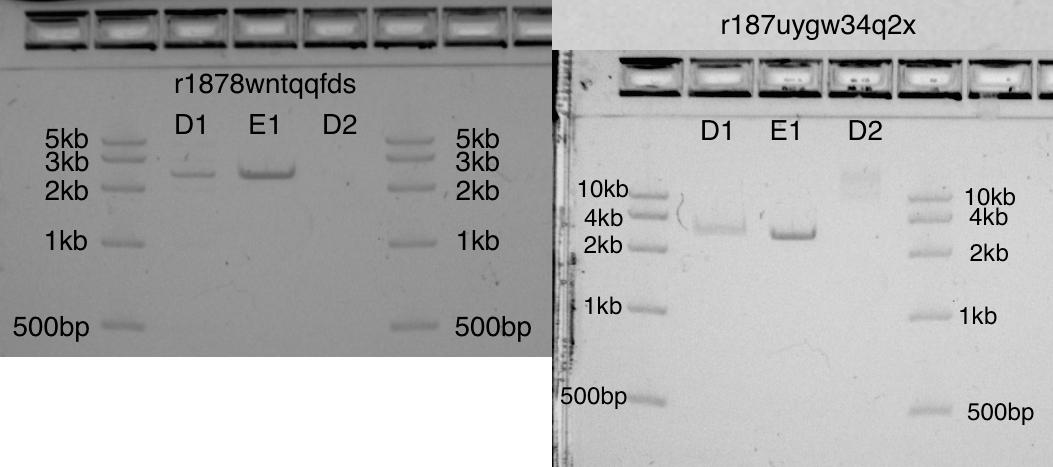 两种凝胶分别在D1和E1带显示了切割的pUC19(2.6kb),在D2中显示了未切割的pUC19
两种凝胶分别在D1和E1带显示了切割的pUC19(2.6kb),在D2中显示了未切割的pUC19步骤3. Gibson组装
, — ,
M13 ( )
qPCR , , . , , , .
, M13 , M13.
代号 """Debugging transformation protocol: Gibson assembly followed by qPCR and a gel v2: include v3 Gibson assembly""" p = Protocol() options = {} experiment_name = "debug_sfgfp_puc19_gibson_seq_v2" inv = { "water" : "rs17gmh5wafm5p",
WARNING:root:Low volume for well sfgfp_puc19_gibson_v1_clone/sfgfp_puc19_gibson_v1_clone : 11.0:microliter
✓ Protocol analyzed
11 instructions
6 containers
Total Cost: $32.09
工作时间:$ 6.98
试剂和消耗品:$ 25.11
结果:用于Gibson组装的qPCR
我可以通过Transcriptic API以JSON格式访问qPCR数据。此功能没有充分的文档记录,但可能非常有用。这些API甚至使您可以访问来自机械手的某些诊断数据,这可以帮助调试。首先,我们要求启动数据: project_id, run_id = "p16x6gna8f5e9", "r18mj3cz3fku7" api_url = "https://secure.transcriptic.com/hgbrian/{}/runs/{}/data.json".format(project_id, run_id) data_response = requests.get(api_url, headers=tsc_headers) data = data_response.json()
然后,我们指定此ID以获取qPCR的“后处理”数据: qpcr_id = data['debug_sfgfp_puc19_gibson_seq_v1_qpcr']['id'] pp_api_url = "https://secure.transcriptic.com/data/{}.json?key=postprocessed_data".format(qpcr_id) data_response = requests.get(pp_api_url, headers=tsc_headers) pp_data = data_response.json()
这是每个管的Ct值(循环阈值)。Ct只是荧光超过特定值的点。她说目前大概有多少DNA(因此大概是我们从哪里开始)。
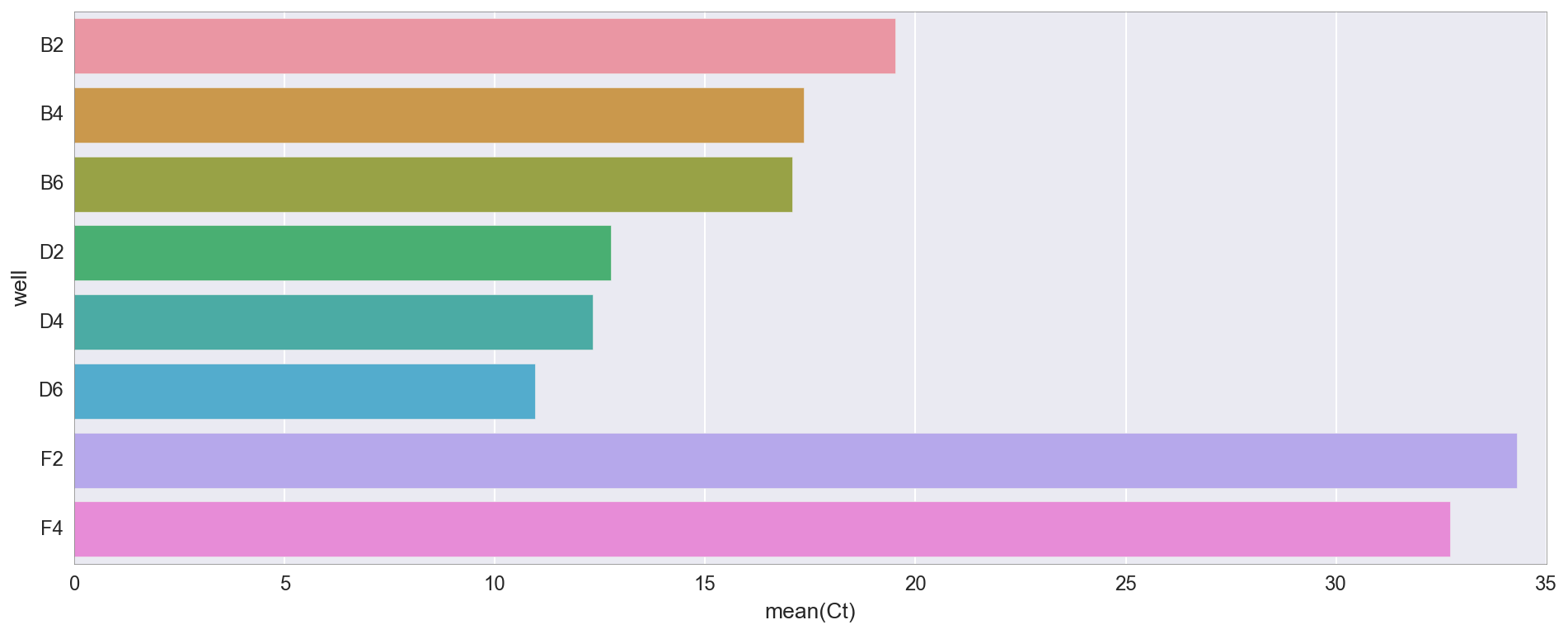
, D2/4/6 ( «v3»), B2/4/6 ( «v1»). v1 v3 , v3 4X NEB, . 30 (F2, F4), -, , .
qPCR, .
f, ax = plt.subplots(figsize=(16,6)) ax.set_color_cycle(['#fb6a4a', '#de2d26', '#a50f15', '#74c476', '#31a354', '#006d2c', '#08519c', '#6baed6']) amp0 = pp_data['amp0']['SYBR']['baseline_subtracted'] _ = [plt.plot(amp0[w_n[well]], label=well) for well in ['B2', 'B4', 'B6', 'D2', 'D4', 'D6', 'F2', 'F4']] _ = ax.set_ylim(0,) _ = plt.title("qPCR (reds=Gibson v1, greens=Gibson v3, blues=control)") _ = plt.legend(bbox_to_anchor=(1, .75), bbox_transform=plt.gcf().transFigure)
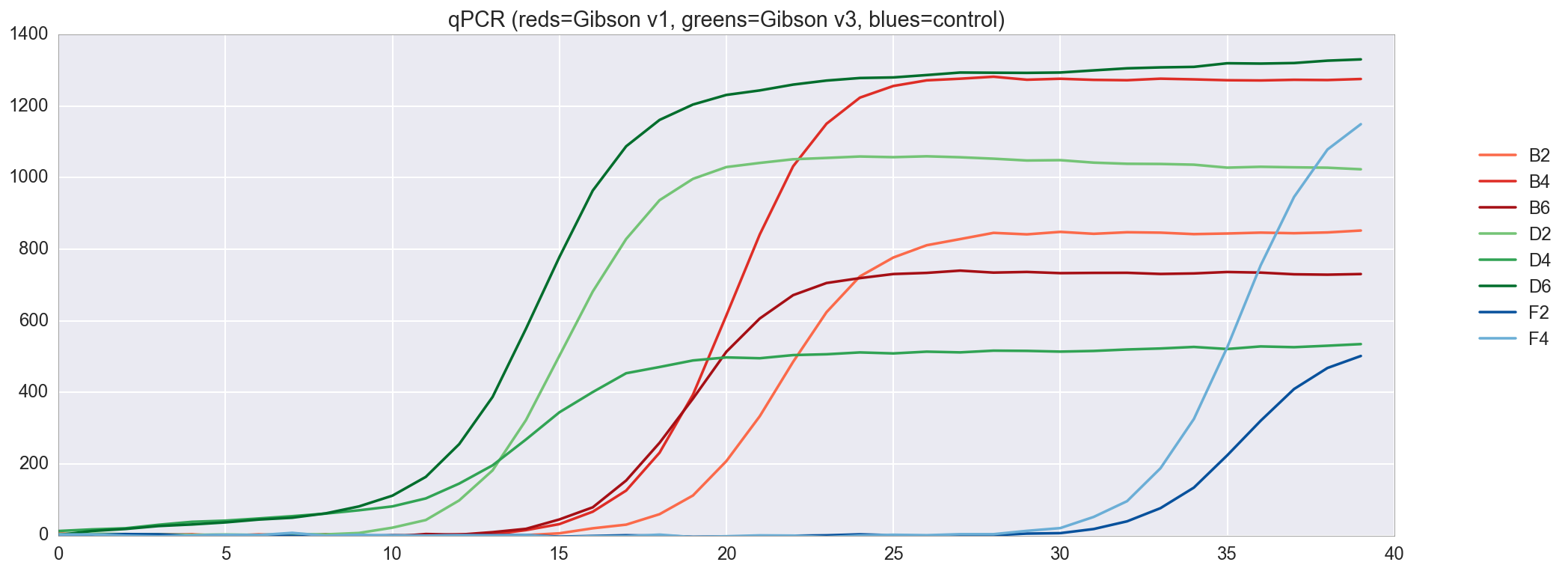
, qPCR , . v3 , v1, .
:
凝胶也非常干净,在B2,B4,B6,D2,D4,D6条带中显示了略低于1kb的强条带:这恰好是我们期望的大小(插入长度约为740bp,M13引物上下大约40bp)。第二泳道对应于引物。您可以确定这一点,因为F2和F4条带仅包含引物DNA。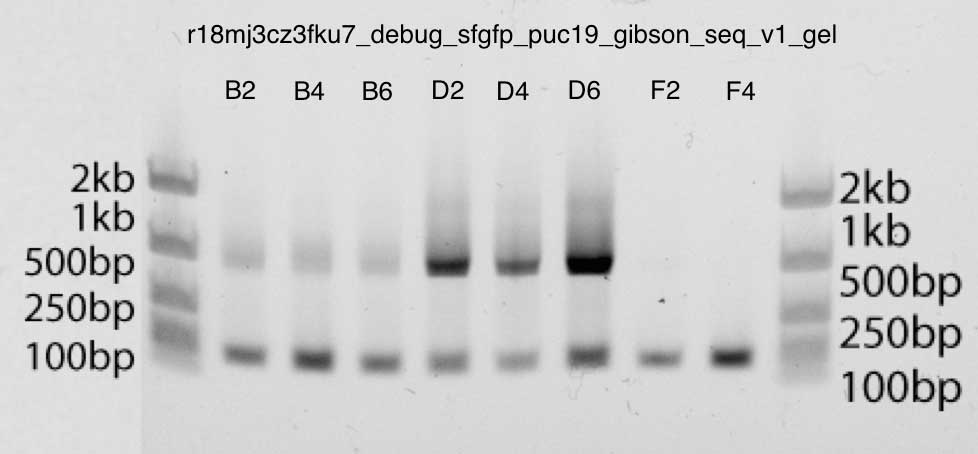 聚丙烯酰胺凝胶电泳:根据上述qPCR数据,c吉布森装配v3显示更强的条带(D2,D4,D6)
聚丙烯酰胺凝胶电泳:根据上述qPCR数据,c吉布森装配v3显示更强的条带(D2,D4,D6)步骤4.转换
转化是通过添加DNA改变身体的过程。在此实验中,我们使用表达sfGFP的质粒pUC19 转化 大肠杆菌。我们使用易于使用的ZymoDH5αMix&Go菌株和推荐的操作方案。该菌株是标准转录本清单的一部分。通常,由于感受态细胞非常脆弱,因此转换可能很复杂,因此协议越简单越可靠,就越好。在普通的分子生物学实验室中,这些感受态细胞对于一般用途而言可能太昂贵了。 具有简单协议的Zymo Mix&Go细胞
具有简单协议的Zymo Mix&Go细胞机器人问题
该协议很好地说明了如何将人工协议适应于机器人的使用是多么困难,以及它可能会意外失败。根据分子生物学家的一般情况,方案有时令人惊讶地含糊(“将管从一侧摆到另一侧”),或者它们可能突然要求进行高级图像处理(“确保沉淀混合在一起”)。人们并不介意这样的任务,但是机器人需要更清晰的说明。. , 37°C. , , , , Transcriptic — , . , , - , . . .
通常有合理的解决方案:有时您只需要使用不同的试剂即可(例如,更多强壮的细胞,例如上述的Mix&Go);有时,您只是以保证金的方式抵押操作(例如,摇动十次而不是三次);有时您需要为机器人设计一些特殊的技巧(例如,使用PCR机进行中暑)。当然,最大的好处是,一旦该协议运行了一次,通常就可以一次又一次地依赖它。您甚至可以量化协议的可靠性,并随着时间的推移对其进行改进!测试转换
, , , pUC19 (. . sfGFP) . pUC19 , , .
(«6-flat» Transcriptic), , . , , , . .
代号 """Simple transformation protocol: transformation with unaltered pUC19""" p = Protocol() experiment_name = "debug_sfgfp_puc19_gibson_v1" inv = { "water" : "rs17gmh5wafm5p",
✓ Protocol analyzed
43 instructions
3 containers
$45.43
:
在下面的照片中,我们看到没有抗生素(左侧板),在所有六个板上均观察到了生长,尽管程度不同,这引起了关注。抄录机器人似乎并不能真正应付均匀分布,这需要一定的技巧。( ) , . , , , , 55 10 . . , . , , .
( , , ,
大肠杆菌。氨苄西林板上的生长要弱得多,尽管那里的细菌比预期的要多得多)。总体而言,尽管存在一些缺陷,但转换的效果足以继续进行。 18小时后用pUC19转化的细胞板:无抗生素(左)和有抗生素(右)
18小时后用pUC19转化的细胞板:无抗生素(左)和有抗生素(右)组装后的产品改造
pUC19, , , , sfGFP.
, IPTG X-gal,
- . , pUC19, sfGFP, .
根据此表,sfGFP在485 nm / 510 nm的激发波长下发光最佳。我发现在Transcriptic中,485/535的效果更好。我想是因为485和510太相似了。我测量600 nm(OD600)的细菌生长。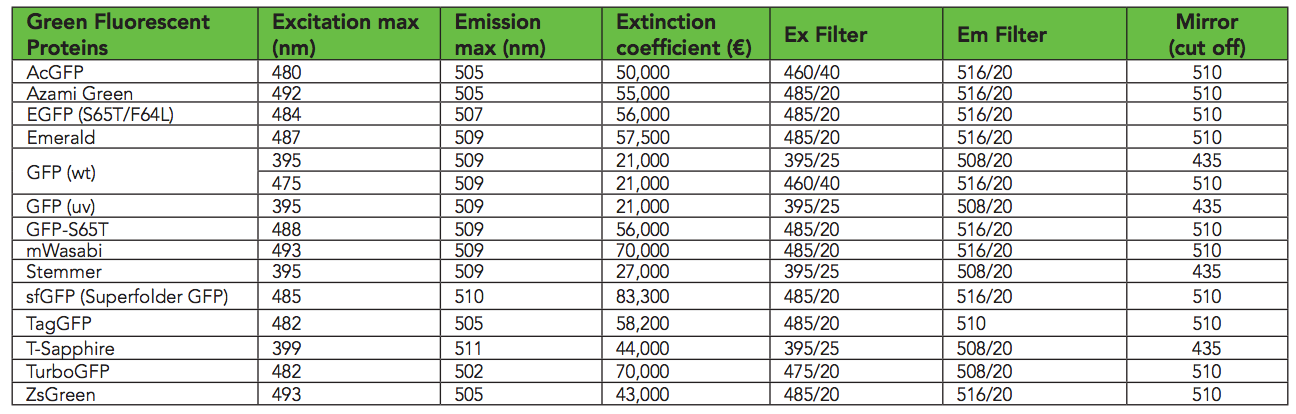 GFP(biotek)品种
GFP(biotek)品种IPTG和X-gal
IPTG 1M 1:1000. , X-gal 20 / 1:1000 (20 /). , 2000µl LB 2 .
40 X-gal 20 / 40 IPTG 0,1 mM ( 4 IPTG 1M), 30 . , IPTG, X-gal .
代号 """Full Gibson assembly and transformation protocol for sfGFP and pUC19 v1: Spread IPTG and X-gal onto plates, then spread cells v2: Mix IPTG, X-gal and cells; spread the mixture v3: exclude X-gal so I can do colony picking better v4: repeat v3 to try other excitation/emission wavelengths""" p = Protocol() options = { "gibson" : False,
Inventory: IPTG/IPTG/IPTG/IPTG/IPTG/IPTG 832.0:microliter {}
Inventory: sfgfp_puc19_gibson_v3_clone/sfgfp_puc19_gibson_v3_clone/sfgfp_puc19_gibson_v3_clone/sfgfp_puc19_gibson_v3_clone/sfgfp_puc19_gibson_v3_clone 57.0:microliter {}
✓ Protocol analyzed
40 instructions
8 containers
Total Cost: $53.20
工作时间:$ 17.35
试剂和消耗品:$ 35.86 殖民地集合
当菌落在氨苄青霉素平板上生长时,我可以“收集”单个菌落并将它们种植在96管平板上。为此,在auto-protocol中有一个特殊的命令(autopick)。代号 """Pick colonies from plates and grow in amp media and check for fluorescence. v2: try again with a new plate (no blue colonies) v3: repeat with different emission and excitation wavelengths""" p = Protocol() options = {} for k, v in list(options.items()): if v is False: del options[k] experiment_name = "sfgfp_puc19_gibson_pick_v3" def plate_expid(val): """refer to the previous plating experiment's outputs""" plate_exp = "sfgfp_puc19_gibson_plates_v4" return "{}_{}".format(plate_exp, val)
✓协议分析
62条指令
8个容器
总成本:66.38美元
工作时间:$ 57.59
试剂和消耗品:$ 8.78
结果:菌落收获
– , , (1-4) (5-6). , , , , IPTG X-gal, Transcriptic.
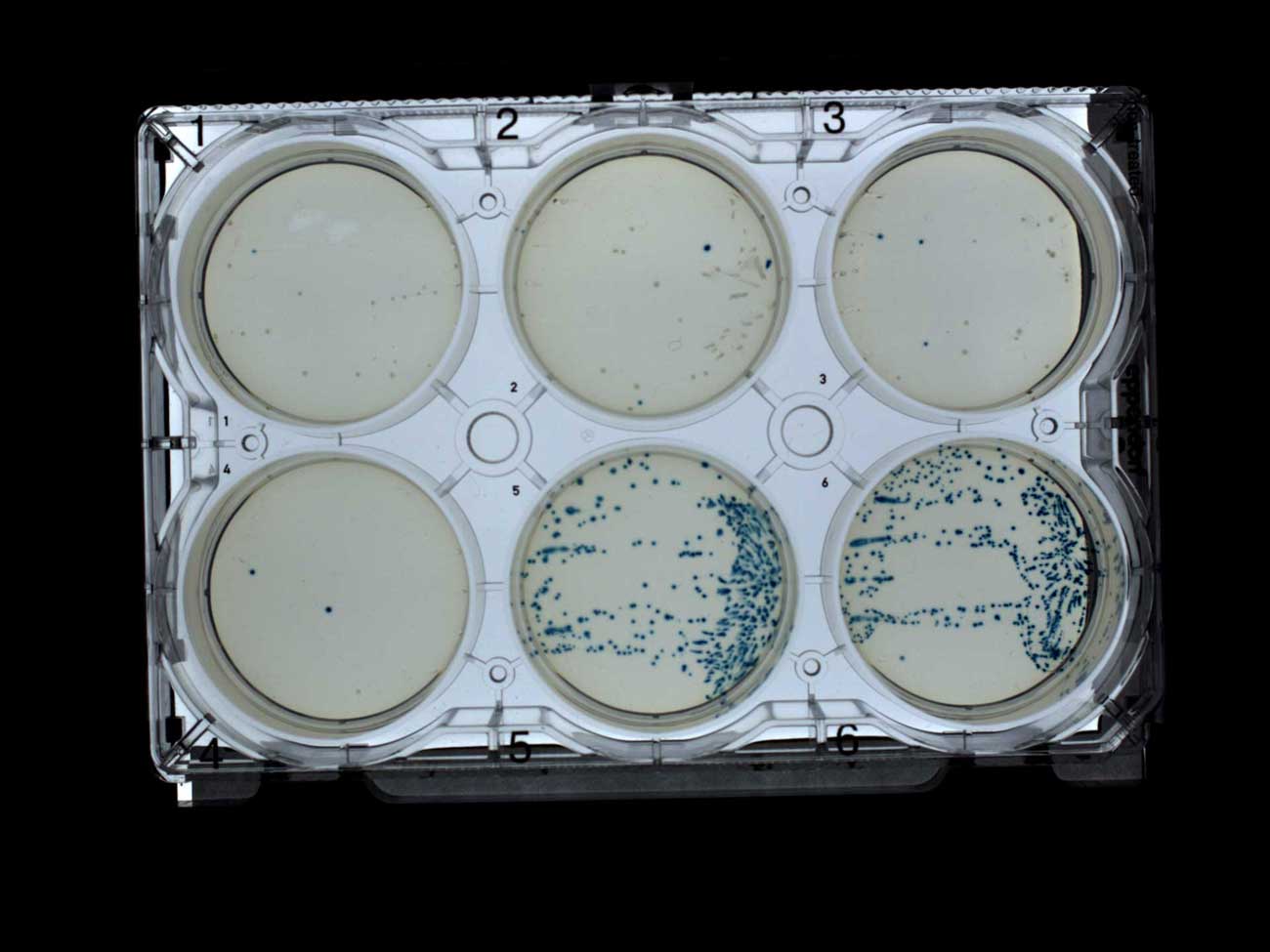 - (1-4) (5-6)
- (1-4) (5-6)- - . (
GraphicsMagick ). , , ( , ).
我还用Transcriptic机器人收集的菌落数量在图像上签名。假定他将从前五个板中最多收集10个菌落。但是,通常会收集到几个菌落,这些菌落通常是蓝色菌落。机器人只能在只有蓝色菌落的控制板上找到十个菌落。我的工作原理是,菌落收集机器人最好收集蓝色菌落,因为它们之间的对比更大。 有氨苄青霉素(1-4)和无抗生素(5-6)的蓝白选择筛板,表明收集的菌落数蓝白两色筛选具有特定目的。他表明大多数殖民地正确地转化了。至少有一个插入。但是,为了更好地收集菌落,我在没有X-gal的情况下重复了实验。只有白色菌落,机器人收集器才能从前五个培养皿的每一个中成功组装出十个菌落。可以假设在大多数收集的菌落中都有成功的插入。
有氨苄青霉素(1-4)和无抗生素(5-6)的蓝白选择筛板,表明收集的菌落数蓝白两色筛选具有特定目的。他表明大多数殖民地正确地转化了。至少有一个插入。但是,为了更好地收集菌落,我在没有X-gal的情况下重复了实验。只有白色菌落,机器人收集器才能从前五个培养皿的每一个中成功组装出十个菌落。可以假设在大多数收集的菌落中都有成功的插入。 在含有氨苄西林(1-4)而无抗生素(5-6)的平板上生长的菌落
在含有氨苄西林(1-4)而无抗生素(5-6)的平板上生长的菌落结果:组装产品的改造
在96管平板上培养了50个选定的菌落20小时后,我测量了荧光以检查sfGFP的表达。 Transcriptic使用Tecan Infinite读取器来测量荧光和吸收(以及发光,如果您愿意的话)。, , , sfGFP. , , - , sfGFP . , sfGFP, , , , .
(OD600) 20 ( 60 ).
for t in [0,4,8,12,16,20]: abs_data = pd.read_csv("glow/sfgfp_puc19_gibson_pick_v3_abs_{}.csv".format(t), index_col="Well") flr_data = pd.read_csv("glow/sfgfp_puc19_gibson_pick_v3_fl2_{}.csv".format(t), index_col="Well") if t == 0: new_data = abs_data.join(flr_data) else: new_data = new_data.join(abs_data, rsuffix='_{}'.format(t)) new_data = new_data.join(flr_data, rsuffix='_{}'.format(t)) new_data.columns = ["OD 600:nanometer_0", "Fluorescence_0"] + list(new_data.columns[2:])
我们在图表上放置了第20小时的数据和以前的测量结果。实际上,我只对最新数据感兴趣,因为那时应该观察到荧光峰。 svg = [] W, H = 800, 500 min_x, max_x = 0, 0.8 min_y, max_y = 0, 50000 def _toxy(x, y): return W*(x-min_x)/(max_x-min_x), HH*(y-min_y)/(max_y-min_y) def _topt(x, y): return ','.join(map(str,_toxy(x,y))) ab_fls = [[row[0]] + [list(row[1])] for row in new_data.iterrows()]
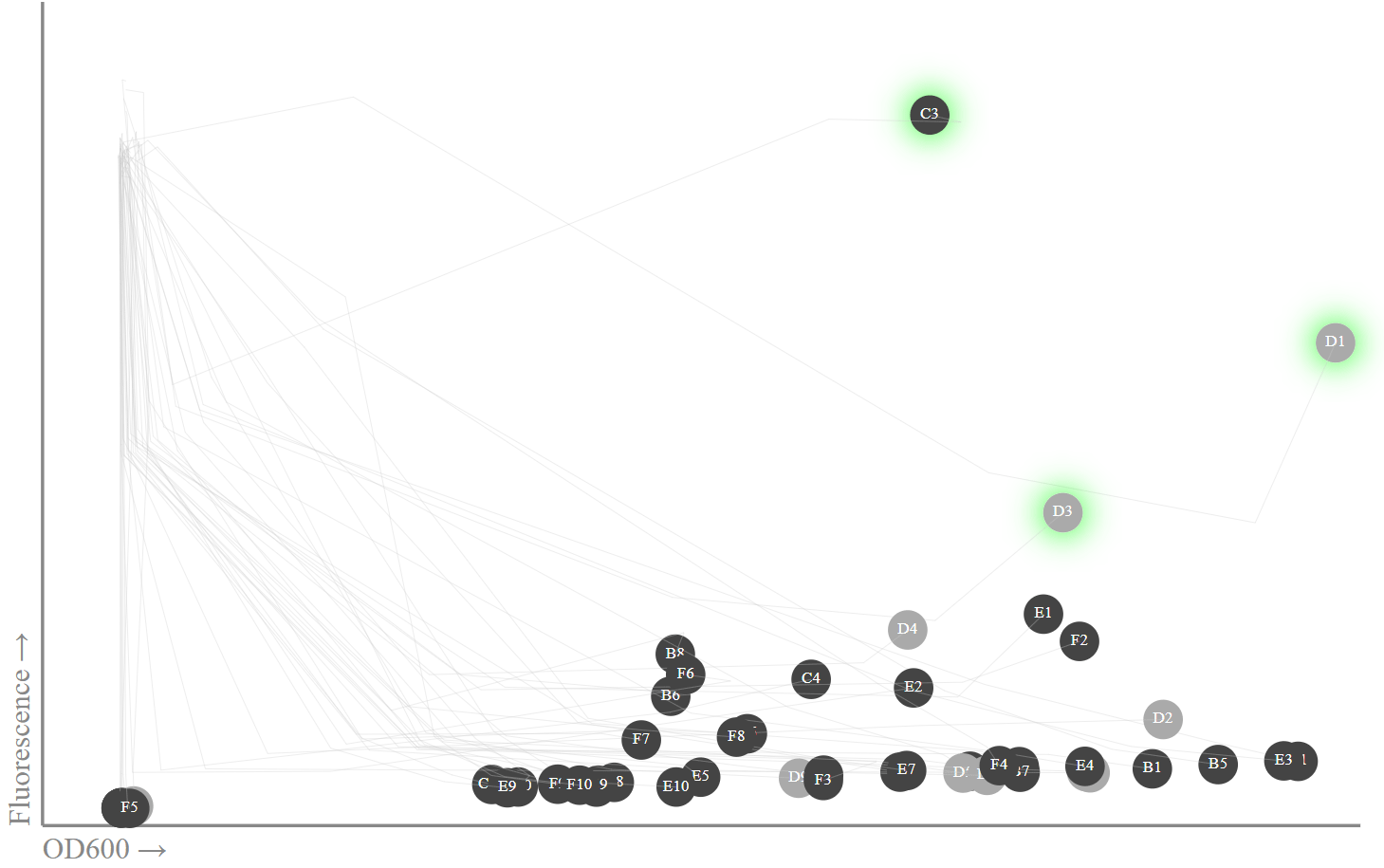 OD600: , . , sfGFPminiprep
OD600: , . , sfGFPminiprep , , 13. , - miniprep - Transcriptic, . (C1, D1, D3) (B1, B3, E1), sfGFP
muscle .
C1, D3 D3 sfGFP, B1, B3 E1 .
, . , 0 (40 000 ). 20- OD600 (, - ), . , , , , 11-15 .
(. . , ), , , ).
根据荧光数据和测序结果,似乎在50个菌落中只有3个产生sfGFP和荧光。这不像我预期的那样多。但是,由于存在三个单独的生长阶段(在板上,体外进行微量制备),因此该细胞的这一阶段已经经历了约200代的生长,因此发生突变的可能性相当大。应该有办法使过程更有效率,尤其是因为我离这些协议的专家还很远。但是,我们仅使用Python代码就成功生产了具有工程GFP表达的转化细胞!第三部分:结论
价钱
根据测量方式的不同,该实验的成本约为360美元,其中不包括调试费:- 70美元的DNA合成
- $32 PCR
- $31
- $32
- $53
- $67
- $75 3 miniprep'
我认为可以通过一些改进将成本降低到$ 250-300。例如,以机器人方式收集50个菌落非常昂贵,而且有可能被废弃。以我的经验,对于某些人(分子生物学家)来说,这个价格似乎很高,而对于其他人(IT人员)来说,这个价格却很低。由于Transcriptic基本上只收取价目表试剂的费用,因此成本的主要区别是人工。这个机器人一个小时已经很便宜了,他不介意半夜起床照相。一旦协议获得批准,很难想象即使是研究生也会更便宜,特别是考虑到机会成本。, . , - , . , : , , IDT .
:
, . , :
- ! , . autoprotocol, .
- . 100 , .
- , , PCR. , , ? / ? , , , « 2-3 ». ?
- . . , .
- . .
- 表现力。您可以使用编程语法来编码重复步骤或分支逻辑。例如,如果要在96管板中分配1至96μl的试剂和(96 − x)μl的水,则可以简单地写出。
- 机器可读数据。具有结果的数据几乎总是以csv格式或其他适合机器处理的格式返回。
- 抽象。理想情况下,无论使用哪种试剂或克隆的样式如何,您都可以运行整个方案,并在必要时替换它(如果效果更好)。
当然,存在一些缺点,特别是因为工具才刚刚开始开发。如果我们将其与Internet进行比较,那么我们将处于1994年:- Transcriptic — . , , , . , , .
- — Transcriptic.
- , . Transcriptic ( , , ).
- 对于许多实验室而言,使用云实验室可能比仅带一名研究生来做这项工作要贵(每小时的边际成本:〜$ 0)。这取决于实验室是否需要研究生的双手或他的智力。
- 周末,Transcriptic尚未进行实验。您可以理解它们,但是即使您的项目很小,也可能不方便。
制作蛋白质的软件
尽管有很多代码和大量调试程序,但我认为可以创建一种将一系列蛋白质作为输入并在输出时表达这种蛋白质的细菌的软件。为此,必须发生几件事:- Twist / IDT / Gen9与Transcriptic的真正集成(由于电流需求低,可能会变慢)。
- , , , , . .
- ( NEB, IDT) (, primer3 ).
(
) , ,
. , in vivo (. . ).
, , : RBS , ; ; .
?
, . :
- - //, .
- , , .
- in vivo split-GFP .
- scFv . scFvs - .
- BiTE , ( , ).
- 制作一种通过毛囊进入人体的局部疫苗(我不建议在家尝试)。
- 以数百种不同的方式对蛋白质进行诱变,看看会发生什么。然后扩展到1000或10,000个突变?也许表征GFP突变?
有关蛋白质设计的新思路,请查看数百个iGEM项目。最后,我要感谢Transcriptic Ben Miles在完成此项目中所提供的帮助。