如何阅读本文 :抱歉,文字如此之长且混乱。 为了节省您的时间,我在每一章的开头都介绍了“我学到了什么”,在其中我用一两个句子解释了本章的本质。
“只是显示解决方案!” 如果您只想了解我的经验,请转至“变得更有创造力”一章,但我认为阅读故障会更有趣和有用。最近,我被指示建立一种处理大量原始DNA序列的方法(从技术上讲,这是SNP芯片)。 必须快速获取给定遗传位置(称为SNP)的数据,以用于后续建模和其他任务。 在R和AWK的帮助下,我能够自然地清理和整理数据,从而极大地加快了请求的处理速度。 这对我来说并不容易,需要多次迭代。 本文将帮助您避免我的一些错误,并演示我最后所做的事情。
首先,一些介绍性的解释。
资料
我们的大学遗传信息处理中心为我们提供了25 TB的TSV数据。 我将它们分成5个由Gzip压缩的软件包,每个软件包包含约240个4 GB的文件。 每行包含一个人的一个SNP的数据。 总共传送了约250万个SNP和约6万人的数据。 除SNP信息外,文件中还有许多列,其编号反映了各种特征,例如读取强度,不同等位基因的频率等。 大约有30列具有唯一值。
目的
与任何数据管理项目一样,最重要的是确定如何使用数据。 在
大多数情况下,
我们将基于SNP选择SNP的模型和工作流程 。 也就是说,与此同时,我们仅需要一个SNP的数据。 我必须学习如何尽可能简单,更快,更便宜地提取与250万个SNP中的一个相关的所有记录。
怎么不做
我会引用一个合适的陈词滥调:
我没有失败过一千次,我只是发现了千种不以方便查询的格式解析一堆数据的方法。
第一次尝试
我了解到 :没有一种便宜的方法可以一次解析25 TB。
在听完范德比尔特大学的“高级大数据处理方法”主题之后,我确定这只是一顶帽子。 配置Hive服务器以运行所有数据并报告结果可能需要一两个小时。 由于我们的数据存储在AWS S3中,因此我使用了
Athena服务,该服务允许您将Hive SQL查询应用于S3数据。 无需配置/提升Hive集群,甚至只需为您要查找的数据付费。
在向Athena显示我的数据及其格式后,我对类似的查询进行了一些测试:
select * from intensityData limit 10;
并迅速获得结构良好的结果。 做完了
直到我们尝试在工作中使用数据...
我被要求提取所有SNP信息以对其进行测试。 我跑了一个查询:
select * from intensityData where snp = 'rs123456';
...并等待。 经过八分钟和超过4 TB的请求数据,我得到了结果。 雅典娜对发现的数据量收取费用,每太字节5美元。 因此,此单个请求的费用为20美元,需要等待八分钟。 要根据所有数据运行该模型,需要等待38年并支付5000万美元,这显然不适合我们。
有必要使用实木复合地板...
我学到的知识 :请注意Parquet文件的大小及其组织。
首先,我试图通过将所有TSV转换为
Parquet文件来纠正这种情况。 它们对于处理大型数据集非常方便,因为其中的信息以列形式存储:每一列都位于其自己的内存/磁盘段中,这与文本文件中的行包含每一列的元素不同。 如果您需要查找某些内容,则只需阅读必要的专栏。 此外,值的范围存储在列中的每个文件中,因此,如果所需的值不在列范围内,Spark将不会浪费时间扫描整个文件。
我运行了一个简单的
AWS Glue任务,将我们的TSV转换为Parquet,并将新文件拖放到Athena中。 花了大约5个小时。 但是,当我启动该请求时,大约花费了相同的时间并花了很少的钱来完成它。 事实是,Spark试图优化任务,只是将一个TSV块拆包并将其放入自己的Parquet块中。 而且由于每个块都足够大并包含许多人的完整记录,因此所有SNP都存储在每个文件中,因此Spark必须打开所有文件以提取必要的信息。
奇怪的是,Parquet中的默认(推荐)压缩类型-snappy-是不可拆分的。 因此,每个执行者都坚持解压缩并下载完整的3.5 GB数据集的任务。

我们了解问题
我学到的是 :排序很困难,尤其是如果数据是分布式的。
在我看来,现在我已经了解了问题的实质。 我要做的就是按SNP列而不是按人员对数据进行排序。 然后,将几个SNP存储在单独的数据块中,然后Parquet智能功能“仅当值在范围内时才打开”将在其所有荣耀中体现出来。 不幸的是,事实证明,整理分布在整个集群中的数十亿行是一项艰巨的任务。
AWS肯定不希望归还这笔钱,原因是“我是一个心不在a的学生。” 在我开始对Amazon Glue进行排序后,它工作了2天并崩溃了。
分区呢?
我了解到 :Spark中的分区应该保持平衡。
然后我想到了在染色体上划分数据的想法。 其中有23种(还有线粒体DNA和未映射区域的少数)。
这将使您可以将数据分成较小的部分。 如果仅在Glue脚本中的Spark导出函数中添加一行
partition_by = "chr"行,则应将数据分类到存储桶中。
 基因组由许多称为染色体的片段组成。
基因组由许多称为染色体的片段组成。不幸的是,这没有用。 染色体的大小不同,因此信息量也不同。 这意味着Spark发送给工作人员的任务不平衡且执行缓慢,因为某些节点较早完成且处于空闲状态。 但是,任务已完成。 但是,当请求一个SNP时,不平衡再次导致了问题。 在较大染色体上(即我们要从中获取数据的地方)处理SNP的成本仅降低了约10倍。 很多,但还不够。
如果您划分为更小的分区?
我了解到的 :绝对不要尝试做250万个分区。
我决定去散散步,划分每个SNP。 这样可以保证分区大小相同。
坏是一个想法 。 我利用了Glue并添加了无辜的
partition_by = 'snp' 。 任务开始并开始运行。 一天后,我检查并发现到目前为止S3中什么都没写,所以我终止了该任务。 看来,Glue正在将中间文件写入S3中的隐藏位置,以及许多文件,可能是数百万个。 结果,我的失误花费了上千美元,并没有使我的导师满意。
分区+排序
我了解到 :排序仍然很困难,就像设置Spark一样。
进行分区的最后一次尝试是我对染色体进行了分区,然后对每个分区进行了排序。 从理论上讲,这将加快每个请求的速度,因为所需的SNP数据应在给定范围内的几个Parquet块内。 las,对已分区的数据进行排序已被证明是一项艰巨的任务。 结果,我切换到自定义群集的EMR,并使用八个功能强大的实例(C5.4xl)和Sparklyr创建了更灵活的工作流程...
# Sparklyr snippet to partition by chr and sort w/in partition # Join the raw data with the snp bins raw_data group_by(chr) %>% arrange(Position) %>% Spark_write_Parquet( path = DUMP_LOC, mode = 'overwrite', partition_by = c('chr') )
...但是,该任务仍未完成。 我以各种方式进行了调整:我为每个查询执行程序增加了内存分配,使用了具有大量内存的节点,使用了广播变量,但是每次结果只有一半,最终执行者开始失败,直到一切停止。
我越来越有创造力
我的知识 :有时特殊数据需要特殊解决方案。
每个SNP都有一个位置值。 这是与沿着其染色体的碱基数目相对应的数目。 这是组织数据的好方法。 首先,我想按每个染色体的区域进行分区。 例如,位置1-2000、2001-4000等。 但是问题是SNP在整个染色体上分布不均匀,这就是为什么组的大小会有很大差异的原因。
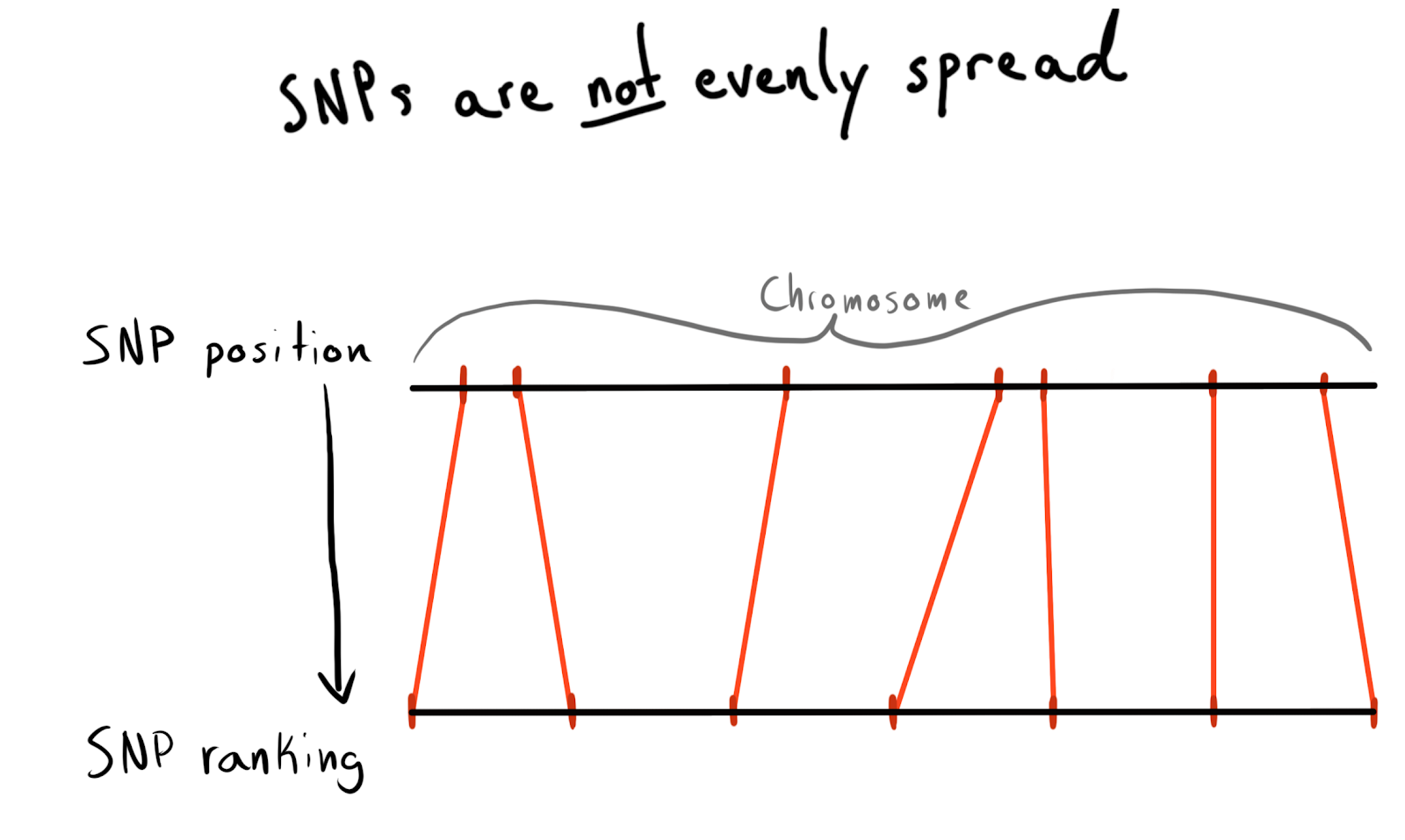
结果,我被分为几类(排名)职位。 根据已经下载的数据,我发出了一个请求,要求提供一个独特的SNP列表,它们的位置和染色体。 然后,他对每个染色体内的数据进行分类,并将SNP收集到给定大小的组(箱)中。 每个说1000 SNP。 这给了我与染色体组的SNP关系。
最后,我在75个SNP上进行分组(bin),下面将解释原因。
snp_to_bin <- unique_snps %>% group_by(chr) %>% arrange(position) %>% mutate( rank = 1:n() bin = floor(rank/snps_per_bin) ) %>% ungroup()
首先尝试使用Spark
我了解到 :Spark集成速度很快,但是分区仍然很昂贵。
我想在Spark中读取这个小数据行(250万行),将其与原始数据合并,然后按新添加的
bin列进行分区。
我使用了
sdf_broadcast() ,因此Spark发现它应该将数据帧发送到所有节点。 如果数据较小且所有任务都需要该数据,则此功能很有用。 否则,Spark会变得聪明并根据需要分配数据,这可能会导致刹车。
再说一次,我的想法没有用:这些任务工作了一段时间,完成了合并,然后,就像通过分区启动的执行器一样,它们开始失败。
添加AWK
我学到的知识 :当基础知识教你时不要睡觉。 当然,早在1980年代就有人解决了您的问题。
到现在为止,我所有Spark失败的原因都是集群中的数据混乱。 也许可以通过预处理来改善这种情况。 我决定尝试将原始文本数据拆分为染色体列,因此我希望为Spark提供“预分区”数据。
我在StackOverflow上搜索了如何分解列值,并找到
了一个很好的答案。 使用AWK,您可以通过写入脚本将文本文件拆分为列值,而不是将结果发送到
stdout 。
为了测试,我编写了一个Bash脚本。 我下载了一个打包的TSV,然后用
gzip解压缩了该文件,并将其发送到
awk 。
gzip -dc path/to/chunk/file.gz | awk -F '\t' \ '{print $1",..."$30">"chunked/"$chr"_chr"$15".csv"}'
奏效了!
芯填充
我学到的 :
gnu parallel是一件神奇的事情,每个人都应该使用它。
分离速度相当慢,当我开始使用
htop测试功能强大(且价格昂贵)的EC2实例的使用时,结果发现我仅使用一个内核和大约200 MB的内存。 为了解决该问题并且不损失很多钱,有必要弄清楚如何并行化工作。 幸运的是,在Jeron Janssens
在Command Line一书中令人惊叹的
Data Science中 ,我找到了有关并行化的章节。 从中我了解了
gnu parallel ,这是在Unix上实现多线程的一种非常灵活的方法。
当我使用新过程启动分区时,一切都很好,但是有一个瓶颈-将S3对象下载到磁盘并不是太快,而且还没有完全并行化。 为了解决这个问题,我这样做:
- 我发现可以直接在管道中实现S3下载步骤,从而完全消除了磁盘上的中间存储。 这意味着我可以避免将原始数据写入磁盘,并且可以使用更小,因此价格更低的AWS存储。
aws configure set default.s3.max_concurrent_requests 50命令aws configure set default.s3.max_concurrent_requests 50大大增加了AWS CLI使用的线程数(默认情况下为10)。
- 我切换到针对网络速度进行了优化的EC2实例,名称中带有字母n。 我发现使用n实例时计算能力的损失可以通过下载速度的提高来弥补。 对于大多数任务,我使用了c5n.4xl。
- 我将
gzip更改为pigz ,这是一个gzip工具,可以做一些很酷的事情来并行化最初无与伦比的解压缩文件任务(这对您的帮助最少)。
这些步骤相互结合,因此一切工作都非常迅速。 由于提高了下载速度,并且拒绝写入磁盘,因此我现在可以在几个小时内处理一个5 TB的软件包。
该推文应该提到“ TSV”。 las
使用重新分析的数据
我了解到的是 :Spark喜欢未压缩的数据,并且不喜欢合并分区。
现在,数据以解压缩(读取,共享)和半排序格式存储在S3中,我可以再次返回Spark。 一个惊喜在等待着我:我再次未能达到期望! 很难确切地告诉Spark如何对数据进行分区。 甚至当我这样做时,事实证明分区太多(9.5万个),当我通过
coalesce将分区的数量减少到一致的极限时,它破坏了我的分区。 我确定可以解决此问题,但是经过几天的搜索,我找不到解决方案。 最后,尽管花了一些时间,但我还是完成了Spark中的所有任务,而且拆分的Parquet文件不是很小(〜200 Kb)。 但是,数据是需要的地方。
 太小了,与众不同,太棒了!
太小了,与众不同,太棒了!测试本地Spark请求
我了解到 :Spark在解决简单问题上有太多开销。
通过以智能格式下载数据,我能够测试速度。 我在R上设置了一个脚本来启动本地Spark服务器,然后从Parquet组(bin)的指定存储库中加载了Spark数据帧。 我试图加载所有数据,但无法让Sparklyr识别分区。
sc <- Spark_connect(master = "local") desired_snp <- 'rs34771739' # Start a timer start_time <- Sys.time() # Load the desired bin into Spark intensity_data <- sc %>% Spark_read_Parquet( name = 'intensity_data', path = get_snp_location(desired_snp), memory = FALSE ) # Subset bin to snp and then collect to local test_subset <- intensity_data %>% filter(SNP_Name == desired_snp) %>% collect() print(Sys.time() - start_time)
执行耗时29.415秒。 好得多,但对于大量测试任何内容都不太好。 此外,我无法使用缓存来加快工作速度,因为当我尝试在内存中缓存数据帧时,即使为重量小于15的数据集分配了超过50 GB的内存,Spark始终会崩溃。
返回AWK
我了解到 :AWK关联数组非常有效。
我知道我可以达到更高的速度。 我记得在
Bruce Barnett出色的AWK指南中,我读到了一个很酷的功能,称为“
关联数组 ”。 实际上,这些是键值对,由于某种原因在AWK中被称为不同的值,因此我不知何故没有特别提及它们。
罗曼·切普利亚卡(Roman Cheplyaka)回忆说,术语“关联数组”比术语“键/值对”要古老得多。 即使您
在Google Ngram中搜索键值 ,您也不会在此处看到该术语,但是会找到关联数组! 另外,键值对最常与数据库关联,因此与哈希图进行比较更具逻辑性。 我意识到我可以使用这些关联数组将我的SNP连接到bin表和原始数据,而无需使用Spark。
为此,在AWK脚本中,我使用了
BEGIN块。 这是一段代码,在第一行数据传输到脚本主体之前执行。
join_data.awk BEGIN { FS=","; batch_num=substr(chunk,7,1); chunk_id=substr(chunk,15,2); while(getline < "snp_to_bin.csv") {bin[$1] = $2} } { print $0 > "chunked/chr_"chr"_bin_"bin[$1]"_"batch_num"_"chunk_id".csv" }
while(getline...)命令加载了CSV组(bin)中的所有行,将第一列(SNP名称)设置为
bin关联数组的键,将第二个值(group)设置为值。 然后,在应用于主文件各行的
{ }块中,将每一行发送到输出文件,该文件根据其组(bin)而获得唯一的名称:
..._bin_"bin[$1]"_...batch_num和
chunk_id对应于管道提供的数据,避免了竞争状态,并且
parallel启动的每个执行线程都写入其自己的唯一文件。
由于我将所有原始数据分散到上一次使用AWK实验后剩下的染色体上的文件夹中,因此现在我可以编写另一个Bash脚本来一次处理该染色体,并将更深的分区数据提供给S3。
DESIRED_CHR='13'
该脚本有两个
parallel部分。
第一部分从所有包含所需染色体信息的文件中读取数据,然后将这些数据分布在各个流中,这些流将文件分散到相应的组(bin)中。 为了防止在将多个流写入同一文件时发生争用情况,AWK将用于将数据写入文件的文件名传输到不同的位置,例如
chr_10_bin_52_batch_2_aa.csv 。 结果,在磁盘上创建了许多小文件(为此,我使用了TB EBS卷)。
来自第二个
parallel部分的管道经过各个组(bin),并将它们的各个文件与
cat组合为通用CSV,然后将其发送以进行导出。
广播到R?
我了解到的是 :您可以从R脚本访问
stdin和
stdout ,因此可以在管道中使用它。
在Bash脚本中,您可能会注意到以下行:
...cat chunked/*_bin_{}_*.csv | ./upload_as_rds.R... ...cat chunked/*_bin_{}_*.csv | ./upload_as_rds.R... 它将所有串联的组文件(bin)转换为下面的R脚本。
{}是一种特殊的
parallel技术,它将它发送的所有数据直接插入命令本身的指定流中。
{#}选项提供唯一的线程ID,
{%}代表作业插槽号(重复,但绝不同时)。 可以在
文档中找到所有选项的列表
。 #!/usr/bin/env Rscript library(readr) library(aws.s3) # Read first command line argument data_destination <- commandArgs(trailingOnly = TRUE)[1] data_cols <- list(SNP_Name = 'c', ...) s3saveRDS( read_csv( file("stdin"), col_names = names(data_cols), col_types = data_cols ), object = data_destination )
将
file("stdin")变量传递到
readr::read_csv ,转换为R脚本的数据将加载到框架中,然后使用
aws.s3作为
.rds文件直接写入S3。
RDS有点像Parquet的年轻版本,没有多余的列存储空间。
完成Bash脚本后,我收到了位于S3中的
.rds文件,这使我可以使用有效的压缩和内置类型。
尽管使用了制动器R,但一切工作都非常快。 毫不奇怪,R上负责读写数据的片段得到了优化。 在一条中等大小的染色体上进行测试后,该任务在大约两个小时内在C5n.4xl实例上完成。
S3限制
我了解到 :由于路径的智能实现,S3可以处理许多文件。
我担心S3是否可以处理传输到它的许多文件。 我可以使文件名有意义,但是S3将如何查找它们?
 S3 ,
S3 , / . FAQ- S3., S3 - . (bucket) , — .
Amazon, , «-----» . : get-, . , 20 . bin-. , , (, , ). .
?
: — .
: « ?» ( gzip CSV- 7 ) . , R Parquet ( Arrow) Spark. R, , , .
: , .
, .
EC2 , ( , Spark ). , , AWS- 10 .
R .
S3 , .
library(aws.s3) library(tidyverse) chr_sizes <- get_bucket_df( bucket = '...', prefix = '...', max = Inf ) %>% mutate(Size = as.numeric(Size)) %>% filter(Size != 0) %>% mutate(
, , ,
num_jobs , .
num_jobs <- 7
purrr .
1:1000 %>% map_df(shuffle_job) %>% filter(sd == min(sd)) %>% pull(data) %>% pluck(1)
, . Bash-
for . 10 . , . , .
for DESIRED_CHR in "16" "9" "7" "21" "MT" do
:
sudo shutdown -h now
… ! AWS CLI
user_data Bash- . , .
aws ec2 run-instances ...\ --tag-specifications "ResourceType=instance,Tags=[{Key=Name,Value=<<job_name>>}]" \ --user-data file://<<job_script_loc>>
!
: API .
- . , . API .
.rds Parquet-, , . R-.
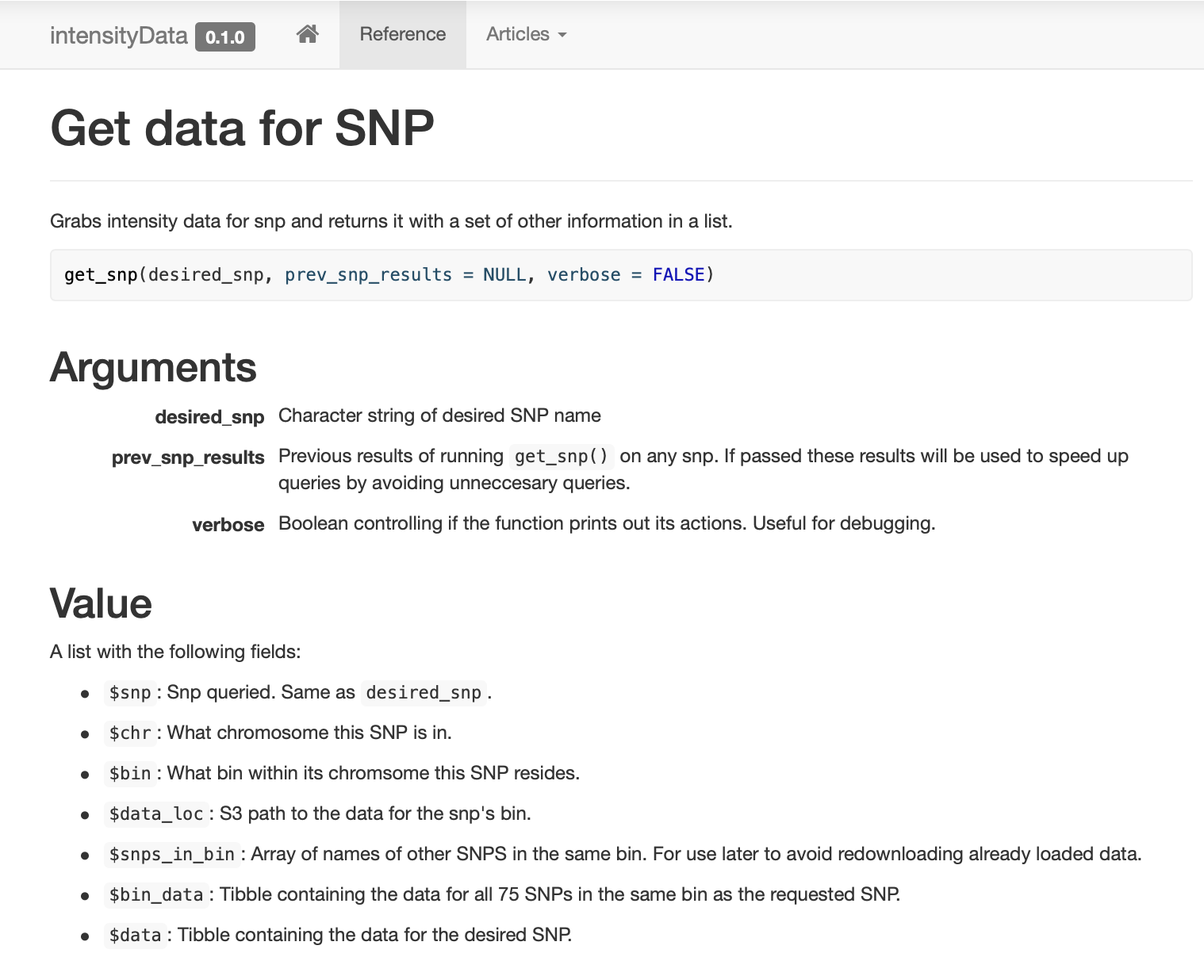
, ,
get_snp .
pkgdown , .
: , !
SNP , (binning) . SNP, (bin). ( ) .
, . , . ,
dplyr::filter , , .
,
prev_snp_results snps_in_bin . SNP (bin), , . SNP (bin) :
结果
( ) , . , . .
, , , …
. . ( ), , (bin) , SNP 0,1 , , S3 .
结论
— . , . , . , , , . , , , , . , , , , - .
. , , «» , . .
:
- 25 ;
- Parquet- ;
- Spark ;
- 2,5 ;
- , Spark;
- ;
- Spark , ;
- , , - 1980-;
gnu parallel — , ;
- Spark ;
- Spark ;
- AWK ;
stdin stdout R-, ;
- S3 ;
- — ;
- , ;
- API ;
- , !