在
上一则有关
完整基因组的详细
文章中 ,我们承诺要发布三个问题,并对第一个正确解决所有这三个问题的人进行测试。 同时,我们给出了在这些任务中如何使用遗传数据的示例。 今天,我们发布第一本。

在第一
篇文章中,我们共享了有用的信息和链接,这些信息和链接对于处理生物信息学数据非常有用。 如果您错过了它,我们建议您先阅读它。
免责声明由于某些命令和软件在Windows上不可用,因此在Unix系统(Linux,macOS)上进行遗传数据的工作。 因此,对于Windows用户,最简单的解决方案之一就是租用Linux虚拟机。
下述所有操作均在命令行-终端上执行。 在开始之前,请了解如何在运行操作系统的终端上工作并使用命令,因为其中一些命令可能会损害操作系统和数据。
必备软件
我们已经在Yandex.Cloud上使用所有必需的软件收集
了虚拟机 (VM)的映像。 在Yandex.Cloud中注册,在“计算云”部分的帐户中,单击“创建VM”。 从Atlas数据分析目录中选择1000个基因组作为公共图像。
虚拟机配置:100%2vCPU,8GB RAM,20GB HDD 创建虚拟机时,必须输入支付数据,但不会从帐户中注销任何内容。 在2019年12月31日之前,对代码字的开始和额外授权足以使用Atlas的VM和映像。 要获得完成任务的资助,请将代码字词“ ATLAS”发送给
Yandex.Cloud支持 。
注意:该赠款仅适用于自2019年12月18日以来注册的Yandex.Cloud新用户,或仍具有试用期并具有初始赠款的用户。 代码字ATLAS仅有效一次。
首先,在要从中连接到VM的本地计算机上创建一个ssh密钥:
ssh-keygen -o -t rsa -b 4096 -C "my-local-machine" -f ~/.ssh/yandex-cloud -a 100
创建VM时,不要忘记将
~/.ssh/yandex-cloud.pub文件的内容复制到适当的窗口。
如果要在计算机上安装软件,则下面是所有安装信息。 如果决定使用Yandex.Cloud,请创建一个VM并继续进行下一部分。
链接
Plink是用于处理遗传数据和广泛基因组关联搜索(GWAS)的软件包。 它是由遗传学家肖恩·珀塞尔(Shaun Purcell)开发的。 自2008年以来,在Plink的帮助下,全球范围内已执行了数百项GWAS,Atlas的最佳结果被Atlas用作计算疾病风险的算法的数据源。
Plink提供了一套用于存储和转换基因分型数据并进行搜索的工具。 Plink还允许进行统计处理,连锁不平衡(LD)分析,后裔身份(IBD)和身份(IBS状态)分析,人群分层和上位性测试-几种遗传变异的相互作用在彼此之间。
IBD和IBS用于分析人口组成并确定亲属关系。
上位性的一个例子是APOE基因中的rs7412和rs429358的变异,变异的某种组合会显着增加罹患阿尔茨海默氏病的风险,而每种变异对风险的贡献很小。
从官方
网站下载稳定版的Plink。
BCF工具
BCFtools是一组用于处理VCF格式及其二进制副本BCF的遗传数据的实用程序。 BCFtools软件包的可能应用程序列表包括VCF / BCF文件的注释,过滤,合并和拆分,找到它们的交点,建立索引,选择性搜索,排序,统计计数等。
要安装,请执行以下操作:
git clone git://github.com/samtools/htslib.git git clone git://github.com/samtools/bcftools.git cd bcftools
安装过程将在
此处更详细地描述。
国王
KING程序包(基于亲缘关系的Gwas推理)在处理来自全基因组搜索关联的数据以确定在研究数据中的亲属关系时用于人口研究。 在这项任务中,KING将帮助确定1000个基因组项目中多个样品的亲缘关系。
您可以
在此处下载。 要解决问题,可
在此处找到KING手册。
在Stackoverflow或其生物信息学对立物Biostars上描述了在使用工具期间可能发生的几乎所有错误。
使用数据
作为指导,我们使用
来自 1000个基因组项目的开放数据。 为了进行分析,我们选择了10个样本,这些样本具有通过分析与参考基因组GRCh37版本对齐的NGS数据而获得的约8,500万种变异的基因型信息。 家庭关系和样本人口见图1。
 图1
图1 VCF样本中使用的谱系。 正方形对应于男性,圆圈对应于女性。 虚线表示不确定的二阶血统。
注意事项
如果在生成VCF期间知道某个人的字段信息,则可以使用VCF格式将其存储为单个数字。 看起来像这样:X染色体记录的GT字段(基因型,基因型)包含一个数值,对应于一个等位基因,男性对应两个,女性对应两个。 如果没有有关测序样品的生物学视野的信息,则GT视野将默认包含两个数值(图2中以红色突出显示)。
在本手册中使用的VCF文件中,Y染色体被排除在外,但是VCF文件中存在Y染色体并不总是意味着测序的样品确实具有它。 这是由于伪常染色体区域(PAR),其对于X和Y染色体而言是相同的,并且位于其末端。
不同的染色体通常不具有长的相同(同源)区域,但是,X和Y染色体从一开始(PAR1)和末端(PAR2)就有几百万个碱基对。 因此,在分析PAR区男性的NGS数据时,发现了两个等位基因(每个性染色体一个),而在女性中,Y型的PAR区可能出现了基因型,尽管实际上它们是X染色体的基因型。
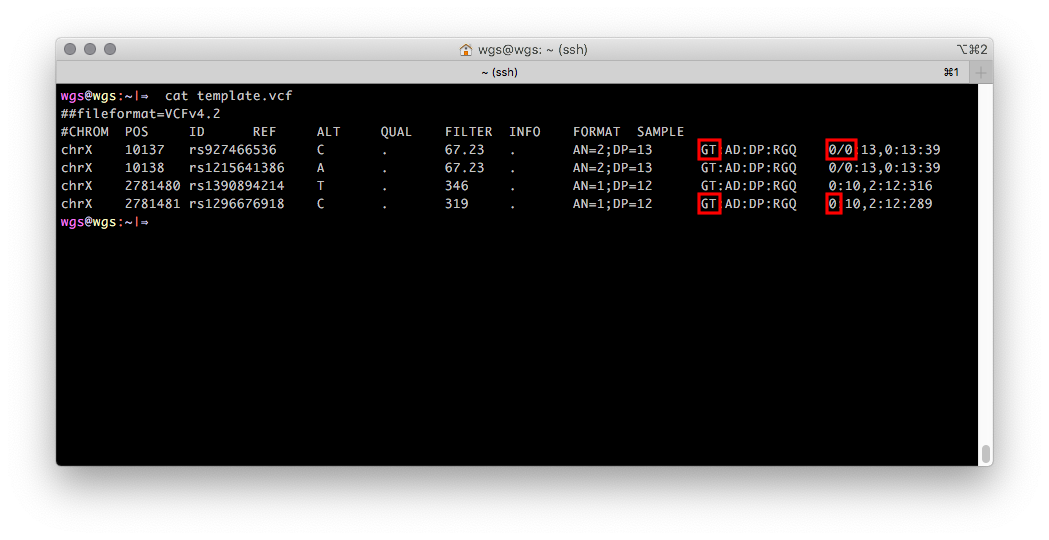 图2
图2 VCF文件,其基因型来自PAR1区(前两个条目)和非伪常染色体区(后两个条目)的人的X染色体。
教育单位
遗传性别是一组与男性或女性类型的主要和次要性特征的表现相对应的性染色体。 通常,男人有一个X染色体和一个Y染色体,而女人有两个X染色体。由于生殖细胞,卵子和精子形成的各种疾病,父母可以生出一个拥有极好的性染色体的孩子,这通常会导致发育障碍主要和次要性特征。
两种最常见的染色体性异常是特纳综合征 (一组X0染色体,即只有一个X染色体)和克莱氏综合征 (一组XXY染色体)。
等位基因是一个或多个位于基因组任何位置的核苷酸,并具有其他核苷酸。 该概念用于描述基因型。 区分参考等位基因和替代基因。 它们全部分别存储在VCF文件的REF和ALT字段中。
确定性别
对于Yandex.Cloud用户使用以下所示的结构,用于完成手动和独立任务的所有数据都存储在Yandex.Cloud上。
Tutorial文件夹包含完成手册所需的VCF文件,独立任务的
Test文件夹。
Technical文件夹包含两个文件,其中列出了遗传变异的标识符:
rsids_for_subsetting.txt使用
rsids_for_subsetting.txt ,用于独立执行的任务,将来在Atlas中获取全基因组测序以将基因分型数据上传至第三方服务时可能需要
external_interpretation_rsids.txt 。 除其他事项外,
Tools文件夹包含任务2和3中使用的两个脚本。
home └── ubuntu ├── Data │ ├── Test │ │ ├── CEI.1kg.2019.test.vcf.gz │ │ └── CEI.1kg.2019.test.vcf.gz.tbi │ └── Tutorial │ ├── CEI.1kg.2019.demo.vcf.gz │ └── CEI.1kg.2019.demo.vcf.gz.tbi ├── Technical │ ├── external_interpretation_rsids.txt │ └── rsids_for_subsetting.txt └── Tools ├── convert_plink_delimiter.sh └── create_23andme.sh
将在Yandex.Cloud VM的
/home目录中创建一个文件夹,其名称对应于VM创建阶段指定的用户名。 通过以下命令将所有内容从
/home/ubuntu目录复制到您的目录中:
cd ~ cp -r /home/ubuntu/* ./
其余的在个人PC上工作时,您可以从
链接下载用于第一个任务的必要文件。 下载的档案支持类似于Yandex.Cloud上使用的文件存储结构:
home └── ubuntu ├── Data │ ├── Test │ │ ├── CEI.1kg.2019.test.vcf.gz │ │ └── CEI.1kg.2019.test.vcf.gz.tbi │ └── Tutorial │ ├── CEI.1kg.2019.demo.vcf.gz │ └── CEI.1kg.2019.demo.vcf.gz.tbi ├── Technical │ ├── external_interpretation_rsids.txt │ └── rsids_for_subsetting.txt └── Tools ├── convert_plink_delimiter.sh └── create_23andme.sh
使用以下命令解压缩
atlas_wgs_contest.tar.gz归档文件
tar -xvzf atlas_wgs_contest.tar.gz 用于以未存档形式执行任务的VCF文件每个占用大约19 GB,因此,为了节省空间,我们建议仅使用存档。 上面列出的所有程序都已经可以使用压缩的VCF数据。 此外,您无需执行任何操作。
为了确定受试者的性别,您需要查看X染色体上的基因型,并排除位于其开始和末端的区域PAR1和PAR2。 这是基因组GRCh37版本中位置60001–2699520和154931044–155260560的间隔。 如果基因型包含一个数字名称,则为男性生物学性别,否则为女性。 应当牢记的是,VCF文件中的性别指定取决于在VCF生成过程中有关生物领域的信息的可用性,因此不能总是使用这种方法。
对数据集中的每个样本使用以下命令。 用
-s参数替换样本标识符:
(/Data/Tutotrial/CEI.1kg.2019.demo.vcf.gz):
执行命令时,您将看到VCF文件中指定样本标识符的部分内容。
-r chrX:2699521-154931043的
-r chrX:2699521-154931043将文件内容的查看限制为从位置2699521到位置154931043(图3中的非PAR区域)的X染色体区域。 这些边界排除了在这种情况下不必要的伪常染色体区域(PAR1和PAR2)。 使用GT字段中的数值,确定每个样品的性别。
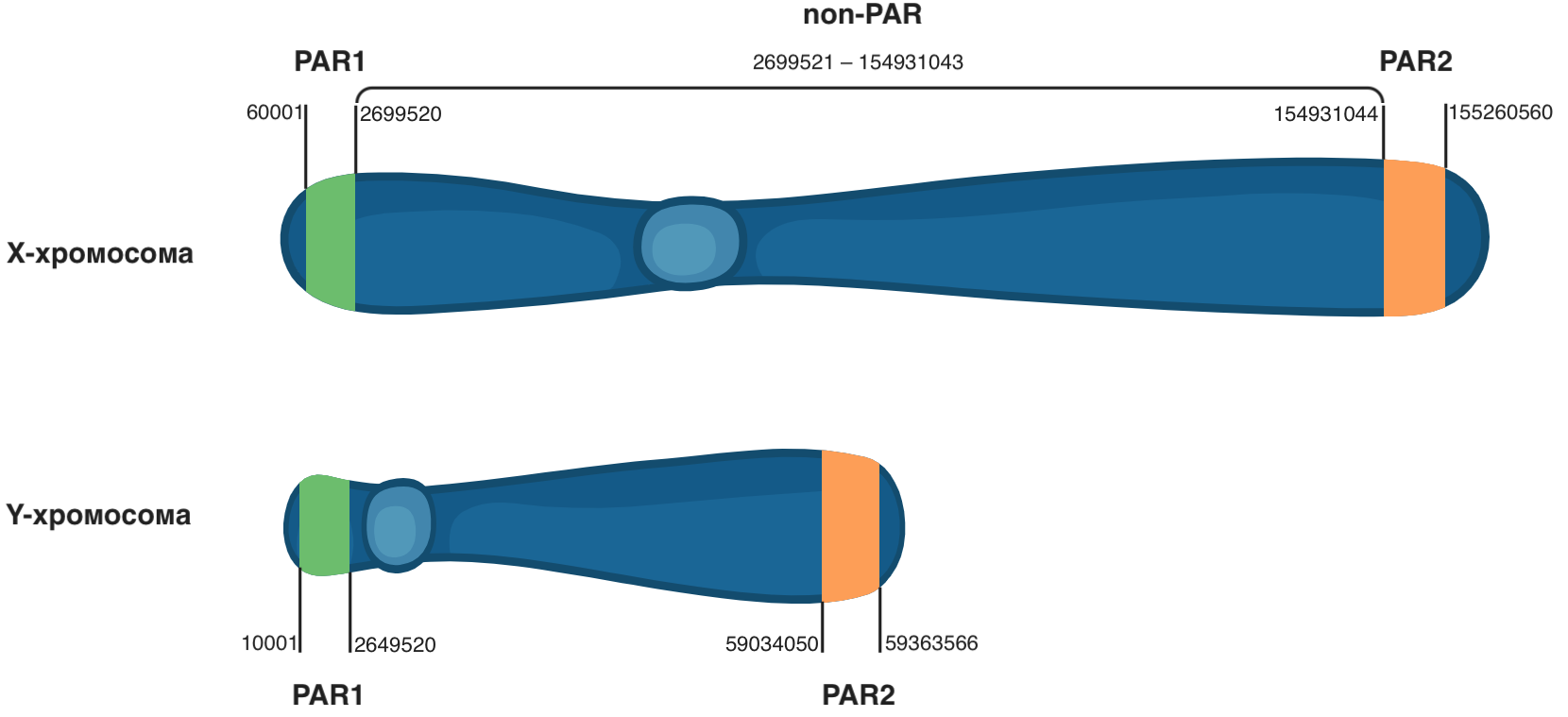 图3
图3 伪常染色体区域PAR1和PAR2在性染色体上的位置。
您可以在图1的VCF文件中或VCF文件标题的最后一行中看到所有样本标识符的列表。 它们将在FORMAT列名称后列出:
这些样品的真实性别也显示在图1中。
我们确定关系
为了确定这种关系,我们需要成对比较所有样品的遗传数据。 根据完整的基因组很难做到这一点:在这种情况下,VCF文件占用数十GB的空间。 我们使用的VCF仅占用约2 GB,但是我们仍然根据Illumina芯片上基因型识别的遗传变异标识符(rsID)列表进行过滤:GSA v1,GSA v2,HumanOmniExpress v1.0,HumanOmniExpress v1.3,InfiniumExome v1。 1和Infinium OmniExpressExome v1.4。 这些是商业基因分型中最受欢迎的芯片。
我们用一个rsID列表在一个单独的文件中汇总了这些芯片的遗传变异所有标识符的列表。 它包含140万个标识符。 要过滤VCF文件,请运行以下命令:
bcftools view -O z -i 'ID=@rsids_for_subsetting.txt' CEI.1kg.2019.demo.vcf.gz > CEI.1kg.2019.demo.subset.vcf.gz
每次您使用BCFtools和其他软件包使用VCF文件时,以前命令的历史记录都会添加到该文件的标题中。 无论使用哪种方法过滤VCF文件和先前执行的命令,都可以通过计算哈希和来检查VCF主要内容的完整性和身份:
gunzip -c命令将文件及其内容输出解压缩到stdout,从中进一步删除
#开头的VCF文件的标题行(因此使用
grep -v "^#"命令)。 删除标头是为了仅比较遗传数据本身的完整性,而不是比较有关哪些工具以及何时使用该VCF文件的元数据。
如果哈希值匹配,则可以继续并将VCF转换为内部Plink格式(默认情况下,Plink格式是三个文件,扩展名为bed,bim和fam)。 在这些文件中,仅保留了基因型,染色体,位置和其他一些数据,其余的被删除了。 使用这种格式,可以更轻松地工作和解决不需要VCF提供其他信息的各种问题。 例如,执行GWAS。
此命令将在文件夹中创建三个文件:
CEI.1kg.2019.demo.subset.bed
CEI.1kg.2019.demo.subset.bim
CEI.1kg.2019.demo.subset.fam您可以确定所有10个样本的成对亲属关系。 我们使用以下命令来分析Plink文件:
king -b CEI.1kg.2019.demo.subset.bed --kinship --prefix CEI.1kg.2019.demo.subset.kinship_analysis
查看文件
CEI.1kg.2019.demo.subset.kinship_analysis.kin0并注意“亲属关系”列,该列包含ID1和ID2中分别指示的成对样本的亲属系数。
将您在文件
CEI.1kg.2019.demo.subset.kinship_analysis.kin0获得的系数与图1所示的谱系进行比较(虚线对应于二阶亲属关系,但是没有确切的亲属关系数据,即可能有堂兄弟,姑姑/侄子或叔叔/侄女。 尝试就亲属系数的哪些值可以对应一阶和二阶相关性做出自己的结论。
提示从KING文档中摘录:亲属系数> 0.354对应于重复的样本或同卵双胞胎,从0.177到0.354对应一阶亲属(父母-子女,兄弟姐妹),从0.0884到0.177对应二阶亲属(堂兄弟,姑姑) /叔叔的侄子),以及从0.0442到0.0884的三级亲属(祖父母,孙子,表亲)。 小于0.0442的任何内容都难以明确解释。
比赛的首要任务
使用包含12个样本的
Data/Test/CEI.1kg.2019.test.vcf.gz的测试数据集,在性别确定和血缘关系分析的结果指导下
Data/Test/CEI.1kg.2019.test.vcf.gz其谱系。 根据分析结果,与某人没有血缘关系的样本应在附近写下,而无需将它们与其他样本连接在一起。 血统书可以采用类似于图1的样式来构成,但是您可以自行决定。 男人用正方形表示,女人用圆圈表示,婚姻用水平线表示,一个孩子用垂直线表示,几个孩子用垂直线的水平分支表示(字母P的形式)。
在此处阅读有关这些名称的更多信息。
如上文所述,亲属系数不能明确地描述一个或另一种亲属的亲属关系:比较父母子女和兄弟姐妹对(一阶亲属)时,可以获得相同的亲属系数。 如果无法确定关系的性质,请指出任何可能的形式。 请注意,测试数据集中的样本具有与训练数据集中使用的标识符不同的标识符。
答复
应发送至
wgs@atlas.ru邮件,直到12月26日至23:59。 即将发布另外两个任务,这些任务的最终结果将在12月28日显示。 获胜者将接受完整基因组测试,第二和第三名将接受Atlas基因测试。
Yandex.Cloud还将特别奖。 Atlas的前任和现任员工均不参加比赛;)