我们继续任务周期,在此讨论如何处理遗传数据。 第一个
任务 “找出性别和关系程度”已经可以解决,并向我们发送答案。 今天,我们发布第二本。
主要奖项是
完整基因组 。

我们以前共享了有用的信息和链接,这些信息和链接可能对处理生物信息学数据有用。 如果您错过了以前的文章,我们建议您先阅读它们:
什么是全基因组,为什么需要任务编号1。 找出性别和关系程度。免责声明
由于某些命令和软件在Windows上不可用,因此在Unix系统(Linux,macOS)上进行遗传数据的工作。 因此,对于Windows用户,最简单的解决方案之一就是租用Linux虚拟机。
下述所有操作均在命令行-终端上执行。 在开始之前,请了解如何在运行操作系统的终端上工作并使用命令,因为其中一些命令可能会损害操作系统和数据。
必备软件
我们已经在Yandex.Cloud上使用所有必需的软件收集
了虚拟机 (VM)的映像。 有关设置虚拟机和安装软件的说明,请参见上
一篇文章第1号任务。
这次,您将需要使用通过主要成分分析方法获得的数据来构建二维散点图。 我们建议您使用任何适合您的软件来构建该图表:Excel,Google表格,Python,R和其他。
要完成任务,您需要Plink 1.9软件包。 如果尚未安装(并且尚未完成任务1),请阅读上一篇文章。 它包含安装说明。 要参加2019年新年大赛,必须完成所有任务!
注意事项
当机器独立搜索数据模式时,主成分分析(PCA)是一种无需教师的机器学习算法。 在遗传学中,PCA允许根据N维空间(通常是二维空间)中的基因分型数据对样本进行聚类,其中获得的主要成分最准确地解释了样本之间遗传数据的可变性。
进行此类分析时,一个总体的样本通常形成一个簇,其边界的大小和平滑度取决于给定总体中样本的相似性。 该算法可能会识别来自不同聚类中不同种群的样本。 如图1所示,来自属于同一超级人口的近距离种群的样本(例如EAS-东亚)将彼此接近甚至在相交的簇中进行识别。
 图1
图1 VCF中使用的样本的谱系(正方形对应于男性,圆圈对应于女性)。 虚线对应于不确定的二阶关系。
类似的分析用于通过基因分型确定种群。 为此,需要一个参考数据集,其中包含具有已知来源的样本。 关于总体的结论可以通过已知样本的哪一组最接近所研究的数据来得出。
为简化起见,PCA分析的本质是多维空间中各点之间的成对距离是已知的,并且这些点必须位于较小的空间中,以使新的成对距离与原始距离成最小程度的差异。 降维可简化数据分析,但我们将其缩减得越多,则点之间的新距离与原始距离的差异就越大。 因此,PCA分析的任务还涉及在准确性和易于分析之间找到折衷方案。 一切都像生活一样。
在遗传数据上执行PCA的最简单变体是基于某些等位基因的身份,这些等位基因可以分为两个亚型:IBS(状态身份)和IBD(血统身份)。 IBS表示两个人中某些等位基因的身份,但不一定暗示它们之间存在任何关系。 相反,IBD则是由于共同祖先的存在以及亲属关系的原因而描述了等位基因的身份。
IBD等位基因无疑是IBS等位基因,反之则不成立。 但是,必须牢记,在某个时间点上我们来自共同祖先,因此某些等位基因可能是IBD。 在下面的PCA分析中,仅使用了IBS概念,尽管在更复杂的分析中它考虑了统计显着性,表型限制,簇大小,年龄和性别以及有关人口结构的其他信息。
两个样本中不同等位基因的数量越多,它们之间的相似性就越小,彼此之间的距离就越远。 这些样本的IBS值会很低。 但是对于父母和他们的孩子来说,IBS将会很高。
了解数据集中每对图像的IBS值后,您可以执行PCA分析以查看它们是如何聚类的。
Atlas遗传测试使用一种更为复杂的算法来确定基因分型数据中的种群代表。
使用数据
我们提醒您,该手册使用了
1000个基因组项目中特别选择的开放数据。 为了进行分析,选择了10个样本,这些样本的基因型信息约为8500万个变异,这些样本是通过分析与GRCh37基因组版本匹配的NGS数据获得的。 这些样品的家庭关系和种群如图1所示。
建立人口集群
使用前面在任务1中获得的Plink格式的三个文件:
CEI.1kg.2019.demo.subset.bed CEI.1kg.2019.demo.subset.bim CEI.1kg.2019.demo.subset.fam
确定训练数据集中所有10个样本之间的成对距离,并根据IBS(状态标识)绘制PCA。 可以按以下步骤完成:
—genome参数仅负责数据集中所有样本之间的IBS / IBD的成对计算。 参数“
—read-genome ”是较早获得的成对距离矩阵,参数“
—cluster —mds-plot 10负责PCA分析,并将其结果输出到10个主要组成部分的表中。 实际上,这些是每个样本在10维空间中的坐标。
最后一个命令将在文件夹中创建4个文件:
CEI.1kg.2019.demo.subset.clustering.cluster1 CEI.1kg.2019.demo.subset.clustering.cluster2 CEI.1kg.2019.demo.subset.clustering.cluster3 CEI.1kg.2019.demo.subset.clustering.mds
我们将需要列表中的最后两个文件。
图2显示了MDS训练数据集上收到的文件的外观。 FID(家庭ID)和IID(个人ID)字段对应于家庭和个体样品标识符。 字段C1-C10包含每个样品的十个主要成分的每个值,其中成分C1最大程度地说明了所分析样品的遗传数据的变异性,而C10则最少。
 图2
图2 MDS文件,每个样本包含10个主要成分的值。
当使用两个分量(在二维空间中)构造散布图时,可以检测与样本总体相对应的聚类。 图3显示了一对主成分C1xC2,C2xC3和C1xC3的散点图。 将获得的聚类与参考种群隶属关系进行比较时(图1),前两个成分C1 – C2对显示最高的准确性(100%),根据1000个基因组计划中声明的种群隶属关系正确分离了所有样本。 但是,由于实际群集可能重叠或分离,因此比较从几对组件中获得的结果总是有意义的。
 图3
图3 成对的主成分的样本位置散点图; 标记的位置略有变化,以防止它们重叠。
使用前三个主要组件构建3D图也可以帮助确定聚类,但并非总是如此。 例如,为图3中的数据构建这样一个图表可以使我们识别出4个聚类,其中PUR和KHV种群的样本根据种群进行了聚类,而CDX种群的样本则分为两个聚类(图4)。 这在图3中的坐标C2xC3和C1xC3中也很明显。
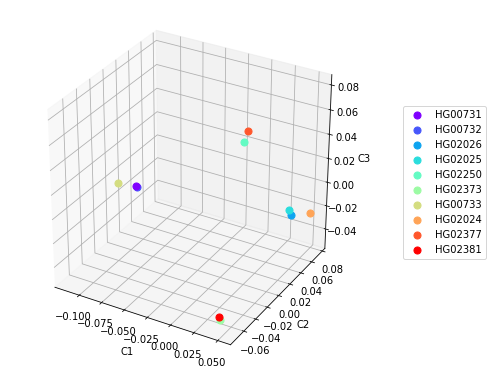 图4
图4 三个主要成分的散点图。
可以通过少量样本来解释这种矛盾的分析结果,因为对于大小和组成不同的数据集,每个样本的主要成分的值都不同,并且当包含来自不同总体的其他样本时,聚类的结果可能会改变。 创建数据集并提供有关样本群体的参考数据时,也有可能发生错误,但是,在1000个基因组计划中,发生这种情况的可能性非常低。
MDS文件不使用制表符或逗号作为分隔符,因此为方便起见,请调整其格式。 使用
tab或
csv作为第二个参数:
该团队将创建
CEI.1kg.2019.demo.subset.clustering.mds.tab文件,您可以下载并构建类似于图3所示的散点图。比较结果,它们应与上面指示的相同。
建立一个聚类树
您还可以使用二叉树评估样本的聚类,该二叉树以离散形式表示有关样本的聚类信息。 关于此树的信息包含在文件
CEI.1kg.2019.demo.subset.clustering.cluster3 (图5)。
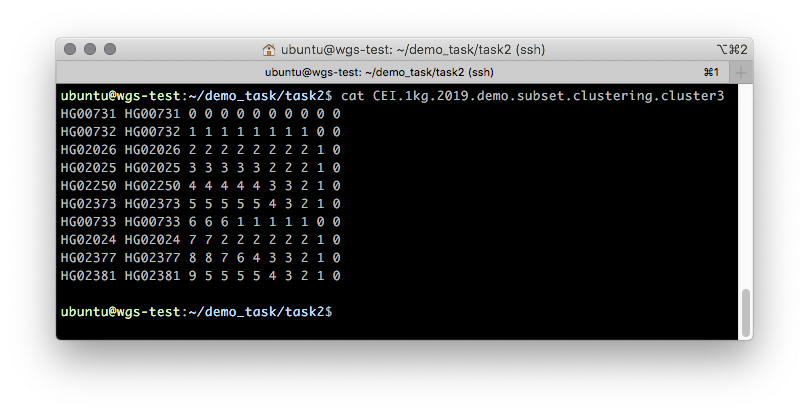 图5
图5 .cluster3文件的大概内容,该文件描述了将样本从1个聚类到N个逐步聚类的过程,其中N是样本数。
该文件的前两列包含FID和IID。 集群隶属关系由其他所有人描述。 该文件应以从右到左的方式以一列的增量在列中读取:最初,所有样本都属于一个簇“ 0”-最右边的列。 当分为两个群集时(第二步,在第二列中),将出现两个群集:“ 0”和“ 1”,其中群集“ 0”包含样本HG00731,HG00732和HG00733,群集“ 1”包含所有其余样本。 图6显示了这种分区。
从树上可以得出结论,样本属于种群(图1)。 此外,这棵树的构建使我们能够建立单个种群的接近度,即将CDX和KHV种群包括在一个EAS超级种群中(已经在拆分超级种群的第一步中,将EAS和AMR分为两个现有分支)。 同样,聚类树的构建可以帮助校正主要组件上的样本可视化的模棱两可的结果。
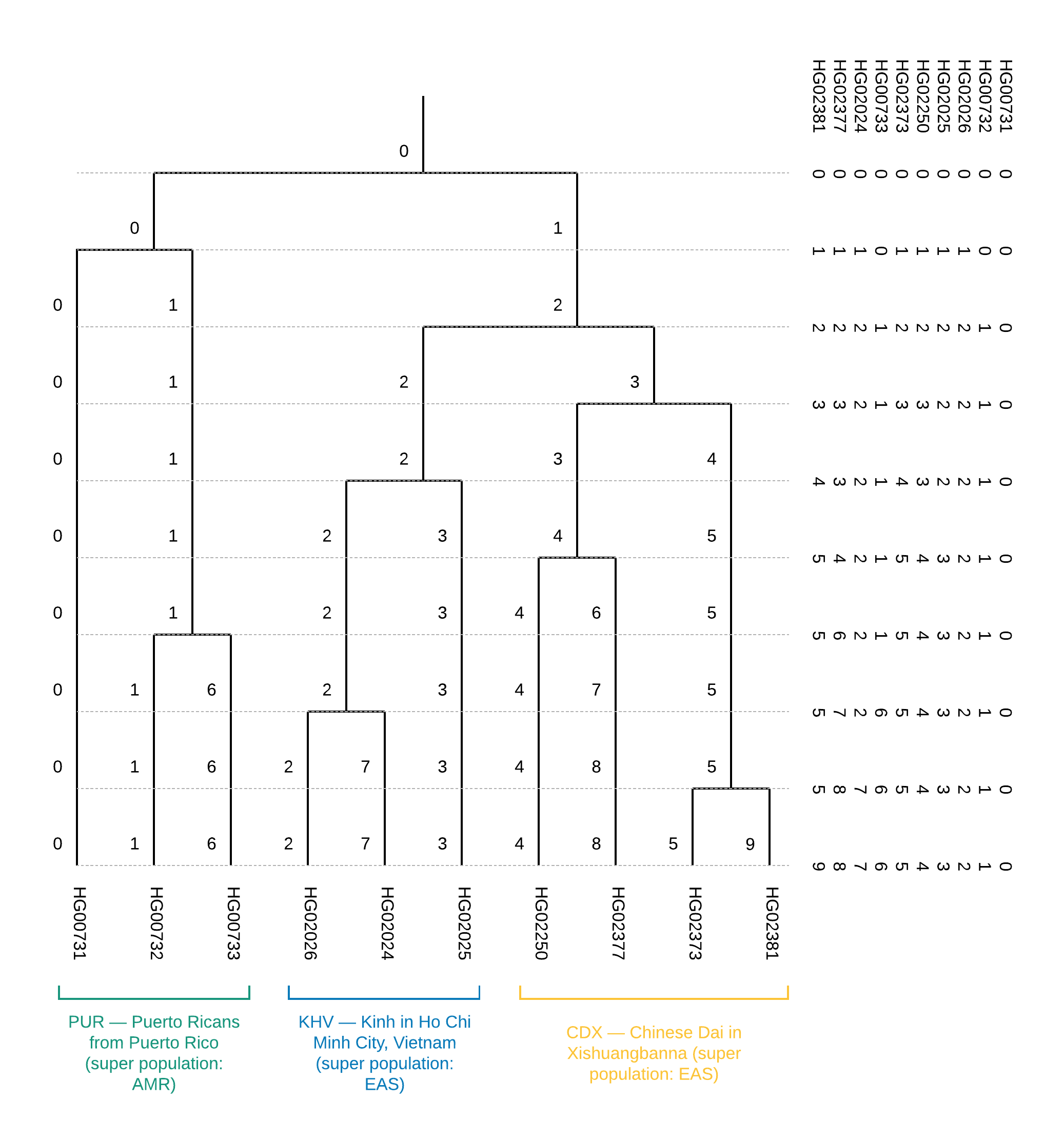 图6
图6 用于包含10个样本的训练数据集的二进制聚类树:右边是文件
CEI.1kg.2019.demo.subset.clustering.cluster3的内容(在文件中从右到左,从上到下相同)。
比赛的第二项任务
使用来自12个样本
Data/Test/CEI.1kg.2019.test.vcf.gz的测试数据集以及上面的示例(图5),从
.cluster3的
.cluster3文件构建二进制聚类树,并将其附加到解决方案中。 分析结果树并得出有关测试数据集中显示的超种群数量的结论。
通过分析主要成分C1,C2和C3,并考虑构建的树,从测试数据集中确定12个样本的总体聚类,并在问题1中构建的谱系中指出这一点,从而限制单个总体块(类似于图1)。 在问题1中未显示亲属关系的样本必须以相同的方式放置在图中获得的块内,而不能将它们与其他样本连接在一起。 别忘了附上您建立的散点图。
答复
应发送至
wgs@atlas.ru邮件,直到12月26日至23:59。 另一个任务将很快发布,最终结果将在12月28日显示。 获胜者将接受完整基因组测试,第二和第三名将接受Atlas基因测试。
Yandex.Cloud还将特别奖。 Atlas的前任和现任员工均不参加比赛;)